
Índice
- Por qué son importantes los factores humanos y las pruebas de usabilidad.
- Incorporación de factores humanos/experiencia universal en la normativa sobre dispositivos médicos.
- Diseño/Evaluación de la Interfaz de Usuario
- Realización de evaluaciones formativas adecuadas
- Pruebas de uso simulado (validación)
- Diseño e interpretación de los resultados de las pruebas sumativas en estudios de validación de uso simulado.
- Factores humanos y pruebas de usabilidad como ventaja competitiva
- Conclusión
- Preguntas Frecuentes (FAQ)
- Recursos y Referencias
La ingeniería de usabilidad, a menudo utilizada indistintamente con los factores humanos, se centra en el diseño de interfaces de usuario (IU) que faciliten una interacción eficiente e intuitiva, un aprendizaje rápido y la satisfacción del usuario. La usabilidad es un concepto multidimensional que describe la facilidad y fiabilidad con la que los usuarios pueden interactuar con un sistema o producto para lograr los resultados previstos. Los factores humanos (FH) y la ingeniería de usabilidad para dispositivos médicos buscan minimizar los riesgos y errores relacionados con el uso para garantizar la seguridad y la eficacia del dispositivo. Las pruebas de usabilidad ya no son opcionales en el desarrollo de dispositivos médicos. Son un factor crítico para la seguridad del paciente, el cumplimiento normativo y el éxito comercial. Las autoridades reguladoras de todo el mundo exigen cada vez más a los fabricantes que demuestren que sus dispositivos pueden ser utilizados de forma segura, eficaz e intuitiva por los usuarios previstos en entornos reales.
A pesar de estas expectativas, muchos fabricantes de dispositivos médicos, en particular las empresas emergentes y las de reciente creación, se enfrentan a dificultades para integrar las actividades relacionadas con los factores humanos en el ciclo de vida del desarrollo de una manera que sea a la vez eficiente y sostenible.
Comprender el propósito, el momento oportuno y el valor regulatorio de las pruebas de factores humanos es esencial para establecer una estrategia que cumpla con la normativa, sea escalable y rentable.
Por qué son importantes los factores humanos y las pruebas de usabilidad.
En las últimas décadas, los eventos adversos han mostrado tendencias preocupantes en los eventos posteriores a la comercialización, atribuibles a problemas de diseño relacionados con la interfaz de usuario (IU) de los dispositivos médicos. Las bombas de infusión, los desfibriladores electrónicos automáticos, los ventiladores y los productos combinados, como los autoinyectores de medicamentos, tienen un historial de problemas de diseño relacionados con el uso que resultan en sobredosis, administración incorrecta, diagnóstico erróneo y retrasos peligrosos en el tratamiento. Como parte del proceso sistemático para reducir errores por parte de los organismos reguladores, las empresas de dispositivos médicos en EE. UU. y la UE se han familiarizado con las disciplinas de Factores Humanos e Ingeniería de Usabilidad (FUS/UE). La FUS/UE se ha aplicado en las industrias automotriz, aeroespacial y de telecomunicaciones durante más de 80 años, pero se integró formalmente en la industria de dispositivos médicos durante la década de 2000, con un importante impulso regulatorio por parte de la FDA en 2011 y una aplicación rigurosa final a partir de 2016.
Las pruebas de factores humanos y usabilidad son cruciales porque garantizan que los productos, en particular los dispositivos médicos y el software, sean seguros, eficaces e intuitivos al evaluar cómo interactúan realmente las personas con ellos. La ingeniería de factores humanos se centra en la interacción entre usuarios, dispositivos y entornos. La usabilidad tiene un gran impacto en la atención médica, especialmente en lo que respecta a la eficacia general de los dispositivos médicos. En pocas palabras, si la usabilidad es deficiente, la realización de las tareas del usuario puede ser más lenta y propensa a errores. Por lo tanto, la administración de la terapia se verá afectada y la seguridad del paciente podría verse comprometida. Es bien sabido que los productos fáciles de usar son más populares, lo que genera discriminación de mercado y ventaja competitiva. Por consiguiente, la usabilidad puede ser un atributo positivo desde una perspectiva comercial y de ventas, además de contribuir al control de riesgos.
Incorporación de factores humanos/experiencia universal en la normativa sobre dispositivos médicos.
En respuesta al creciente número de eventos adversos relacionados con el uso asociados con el diseño de UI, la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) ha integrado las revisiones de HF/UE como un componente rutinario del proceso de aprobación previa a la comercialización. Dentro del Centro de Dispositivos y Salud Radiológica (CDRH), estas revisiones son realizadas por la Oficina de Evaluación y Calidad de Productos (OPEQ) y se guían por la guía publicada por la FDA, Aplicación de factores humanos e ingeniería de usabilidad a dispositivos médicos.
De manera similar, las autoridades reguladoras internacionales han adoptado la norma IEC 62366-1. Dispositivos médicos: aplicación de la ingeniería de usabilidad a dispositivos médicos.Como parte de sus marcos regulatorios fuera de los Estados Unidos, tanto la guía de la FDA como la norma IEC 62366-1 definen un proceso estructurado de ingeniería de usabilidad que abarca el ciclo de vida del desarrollo del dispositivo y culmina en pruebas de validación de usabilidad del diseño final de la interfaz de usuario en condiciones de uso simuladas.
Actividades básicas en el proceso HF/UE y alineación con la evaluación de riesgos del dispositivo.
Los procesos de Ingeniería de Factores Humanos y Usabilidad implican definir grupos de usuarios, realizar análisis de tareas, llevar a cabo evaluaciones formativas y ejecutar pruebas de validación de usabilidad sumativas para garantizar un uso seguro y eficaz del dispositivo. Estas actividades se alinean directamente con el proceso de gestión de riesgos de dispositivos médicos definido por la norma ISO 14971, mediante la identificación, mitigación y validación de controles para riesgos críticos relacionados con el usuario, como los documentados en un Análisis de Riesgos Relacionados con el Uso (URRA).
Los mayores desafíos para los fabricantes de dispositivos
Los siguientes desafíos persisten para cumplir con la intención de la guía de la FDA y la norma IEC 62366-1:
- Realizar evaluaciones formativas adecuadas antes de las validaciones del diseño final.
- Realizar y documentar evaluaciones exhaustivas de riesgos de uso.
- El diseño e interpretación de los resultados de las pruebas sumativas en estudios de validación de uso simulado.
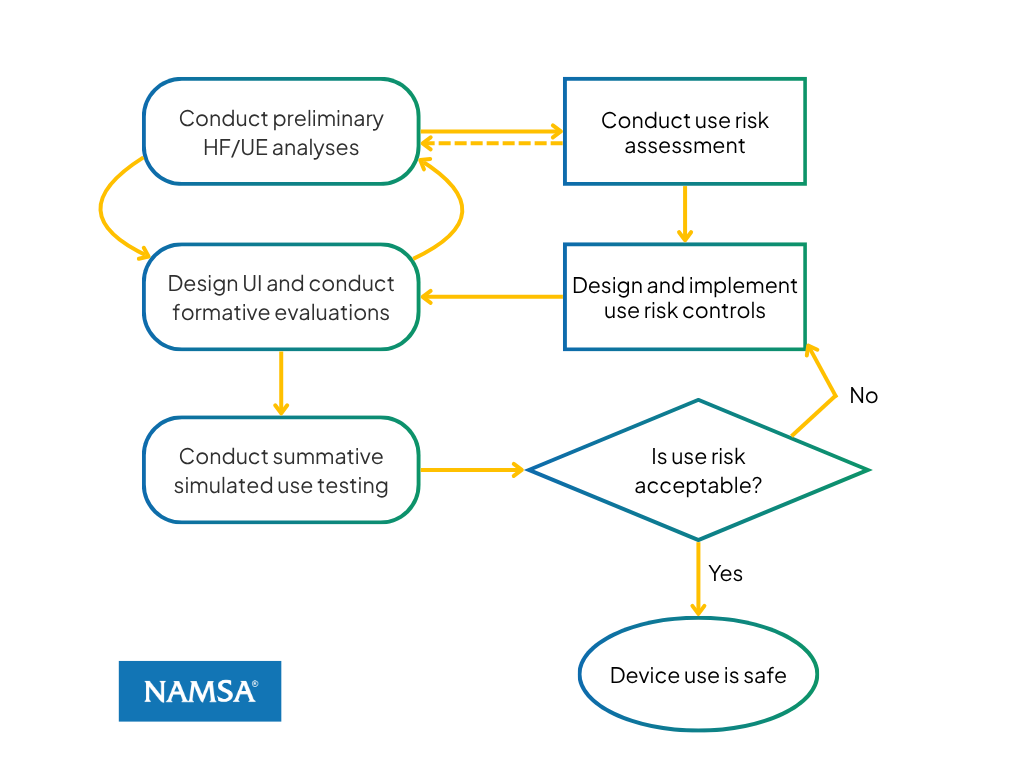
Figura 1: Relación del flujo del proceso HF/UE con el proceso de control de riesgos ISO 14971
La Figura 1 ilustra la relación entre las fases principales de HF/UE y las etapas correspondientes de evaluación y gestión de riesgos de dispositivos. A la izquierda se muestran las fases de las actividades de HF/UE, mientras que a la derecha se muestran las etapas de evaluación y gestión de riesgos, ilustrando cómo se relacionan estos procesos entre sí. La relación entre el proceso HF/UE y la norma ISO 14971 se puede resumir de la siguiente manera:
- Evaluación de RiesgosEl análisis de riesgos según la norma ISO 14971 se apoya en las actividades iniciales de factores humanos/experiencia del usuario, caracterizadas por la realización de un análisis preliminar centrado en la comprensión de los usuarios, su entorno de uso, las tareas de interacción y los posibles peligros relacionados con el uso asociados a la interfaz del dispositivo.
- Implementación del control de riesgosLa implementación de controles de riesgo corresponde al diseño y evaluación de la interfaz de usuario, incluyendo las pruebas de las características de la interfaz destinadas a mitigar los riesgos identificados relacionados con el uso.
- Evaluación del riesgo residualLa aceptación de los controles de riesgo se aborda mediante pruebas de uso simulado, que evalúan si persisten patrones residuales de errores de uso en tareas críticas y si los riesgos relacionados con el uso se han reducido a un nivel bajo aceptable.
Esta alineación garantiza que las actividades de factores humanos/experiencia del usuario estén plenamente integradas en el marco general de gestión de riesgos del dispositivo y proporcionen evidencia objetiva que respalde la seguridad de uso.
Diseño/Evaluación de la Interfaz de Usuario
Tanto la FDA como la norma IEC 62366-1 enfatizan un proceso de diseño de interfaz de usuario (UI) basado en evaluaciones formativas iterativas. Estas evaluaciones se realizan desde las primeras etapas y a lo largo del desarrollo del diseño para mejorar la usabilidad y gestionar los riesgos asociados al uso. Las evaluaciones formativas persiguen dos objetivos principales: mejorar la intuición y la facilidad de uso del dispositivo, e identificar y mitigar los posibles riesgos relacionados con su uso. En algunos casos, estos objetivos pueden implicar concesiones, ya que una mayor simplicidad no siempre se ajusta a los requisitos de seguridad.
Varios recursos establecidos respaldan la evaluación formativa durante el desarrollo temprano de la interfaz de usuario del dispositivo. AAMI HE75, Ingeniería de factores humanos – Diseño de dispositivos médicosLa cláusula 9 proporciona orientación práctica sobre métodos de prueba de usabilidad, como recorridos cognitivos, evaluaciones heurísticas y estudios de recorrido y análisis. El anexo D de la norma IEC 62366-1 también describe estas técnicas en el contexto de la ingeniería de usabilidad.
Las normas de diseño de interfaces y las mejores prácticas, como HE75, ofrecen una excelente guía para el diseño de pantallas, controles, interfaces gráficas de usuario (IGI) de software, alarmas, instrumental quirúrgico, instrucciones de uso (IFU) y otros elementos de las IUI. Sin embargo, HE75 destaca la importancia de las pruebas interactivas con usuarios durante el proceso de diseño de la IUI. El papel de las técnicas formativas, como los recorridos cognitivos, las evaluaciones heurísticas y las pruebas de usabilidad con recorrido y explicación, es fundamental para obtener retroalimentación útil de los usuarios para el equipo de diseño, tanto en lo que respecta a la facilidad de uso como a la mitigación efectiva de errores.
Realización de evaluaciones formativas adecuadas
Si bien las evaluaciones formativas suelen requerir un esfuerzo y recursos modestos, muchos fabricantes limitan o evitan estas actividades durante el desarrollo de la interfaz de usuario. Esto se debe a menudo a ideas erróneas comunes, entre las que se incluyen las siguientes:
- Costo y tiempo percibidos: Los estudios formativos suelen ser eficientes y pueden completarse con un número reducido de usuarios representativos. Si se realizan al inicio del proceso de diseño, con tan solo 8 a 10 participantes se puede identificar el 90 % de los fallos de diseño existentes, lo que permite realizar mejoras oportunas que reducen errores y problemas de uso. A medida que el diseño madura, las evaluaciones formativas también pueden servir como pruebas piloto antes de la validación final, lo que ayuda a evitar rediseños costosos o validaciones repetidas debido a problemas de usabilidad sin resolver o deficiencias en el protocolo.
- La obtención de la aprobación del Comité de Ética Institucional provoca retrasos en la presentación de solicitudes: Muchas empresas creen que las pruebas formativas requieren una aprobación exhaustiva por parte de un comité de ética institucional (CEI) en lo que respecta a la seguridad de los participantes. En la mayoría de los casos, las pruebas de usabilidad no presentan riesgo de daño para los participantes (es decir, no se administra ninguna terapia, ya que la atención se centra en la interacción del usuario con la interfaz de usuario). Sin embargo, los fabricantes pueden optar por protegerse de posibles responsabilidades obteniendo la aprobación del CEI, lo que puede implicar revisiones aceleradas, dada la baja complejidad de las pruebas en comparación con los ensayos clínicos.
- Suposiciones sobre la carga administrativa de la documentación: Desde la perspectiva del cumplimiento con la FDA y la norma IEC 62366-1, las evaluaciones formativas no requieren una documentación exhaustiva en las solicitudes de autorización previa a la comercialización. Los revisores de la FDA simplemente desean saber si el fabricante ha realizado pruebas iterativas del producto y si ha hecho todo lo posible por eliminar los defectos de diseño relacionados con la seguridad. Sin embargo, en los últimos años, los revisores de la FDA han solicitado cada vez más registros de evaluación formativa para demostrar una preparación adecuada antes de la validación sumativa de usabilidad, lo que representa un cambio con respecto a las expectativas de presentación anteriores.
Pruebas de uso simulado (validación)
Las pruebas sumativas, también conocidas como pruebas de validación de uso simulado, constituyen la principal fuente de evidencia que respalda la seguridad de uso según la norma IEC 62366-1 y la guía de factores humanos de la FDA. Para evitar problemas de seguridad evidentes para los participantes, las pruebas sumativas pueden realizarse en condiciones de "uso simulado" que representen las condiciones del mundo real.
Las pruebas sumativas difieren de las evaluaciones formativas en su propósito y ejecución. No Concebido como un estudio exploratorio, no busca sugerencias de diseño; más bien, sirve como demostración final de que el dispositivo puede utilizarse de forma segura y eficaz. Los participantes no reciben instrucciones, interrupciones ni correcciones durante la realización de la tarea.
Los escenarios de uso están diseñados para reflejar las secuencias típicas de interacción con el dispositivo y deben incluir todas las tareas identificadas como de alto riesgo en el análisis de riesgos relacionados con el uso. La capacitación y la familiarización se brindan de manera coherente con las condiciones reales esperadas, considerando la retención del aprendizaje y el posible olvido. Las instrucciones de uso están disponibles durante las pruebas, pero los participantes no están obligados a revisarlas a menos que así lo deseen.
Se observa el desempeño del usuario y se clasifica como éxito, fracaso o éxito con dificultad (por ejemplo, vacilación, autocorrección o confusión). Estas observaciones, junto con cualquier fracaso, se documentan para el análisis posterior a la prueba. Se realizan entrevistas posteriores a la prueba para comprender las causas fundamentales de las dificultades o errores observados y para recopilar comentarios de los participantes sobre la complejidad de la tarea.
Los resultados del estudio deben respaldar una conclusión general sobre la seguridad de uso. Esta determinación no se basa en umbrales de éxito cuantitativos predefinidos, sino en si persisten patrones residuales de problemas relacionados con el uso que puedan atribuirse a la interfaz de usuario, el etiquetado o las instrucciones.
Diseño e interpretación de los resultados de las pruebas sumativas en estudios de validación de uso simulado.
El diseño, la realización y la interpretación de las pruebas de uso simulado sumativas representan uno de los aspectos más complejos de la Ingeniería de Factores Humanos y Usabilidad (HF/UE). Estos estudios constituyen la evidencia principal que respalda la seguridad de uso y se distinguen de las evaluaciones de usabilidad convencionales. Su valor reside no solo en el desempeño observado en la tarea, sino también en el análisis posterior a la prueba, necesario para determinar las causas fundamentales de los errores y dificultades de uso.
Tal como se describe en las directrices de la FDA y en la norma IEC 62366-1, las pruebas sumativas proporcionan a los fabricantes una evaluación prospectiva del uso real del dispositivo. Los resultados inesperados o no deseados observados durante las pruebas deben investigarse mediante un enfoque similar al del análisis de eventos adversos posteriores a la comercialización. Por lo tanto, las observaciones en estas pruebas que sean inesperadas o no deseadas se investigan de la misma manera que un evento adverso. El objetivo es identificar las causas fundamentales de los errores que se producen durante las tareas críticas y determinar si estos problemas pueden mitigarse antes de la aprobación regulatoria y la comercialización.
La presentación al proceso previo a la comercialización de la FDA generalmente ha requerido la inclusión de los resultados de las pruebas de validación sumativas, especialmente para los dispositivos de mayor riesgo. Si bien la calidad general de los estudios ha mejorado con el tiempo, se siguen observando varias deficiencias recurrentes:
- Vinculación insuficiente con el análisis de riesgos relacionados con el usuario (URRA): Los protocolos de prueba deben justificarse explícitamente utilizando el URRA (Figura 2 y XNUMX), con escenarios de uso vinculados a tareas críticas desde una perspectiva de riesgo. Las tareas críticas deben estar claramente documentadas, en lugar de ser inferidas implícitamente por el revisor. Una presentación clara del análisis de tareas, los controles de riesgo y las actividades de validación fortalece la revisión regulatoria.
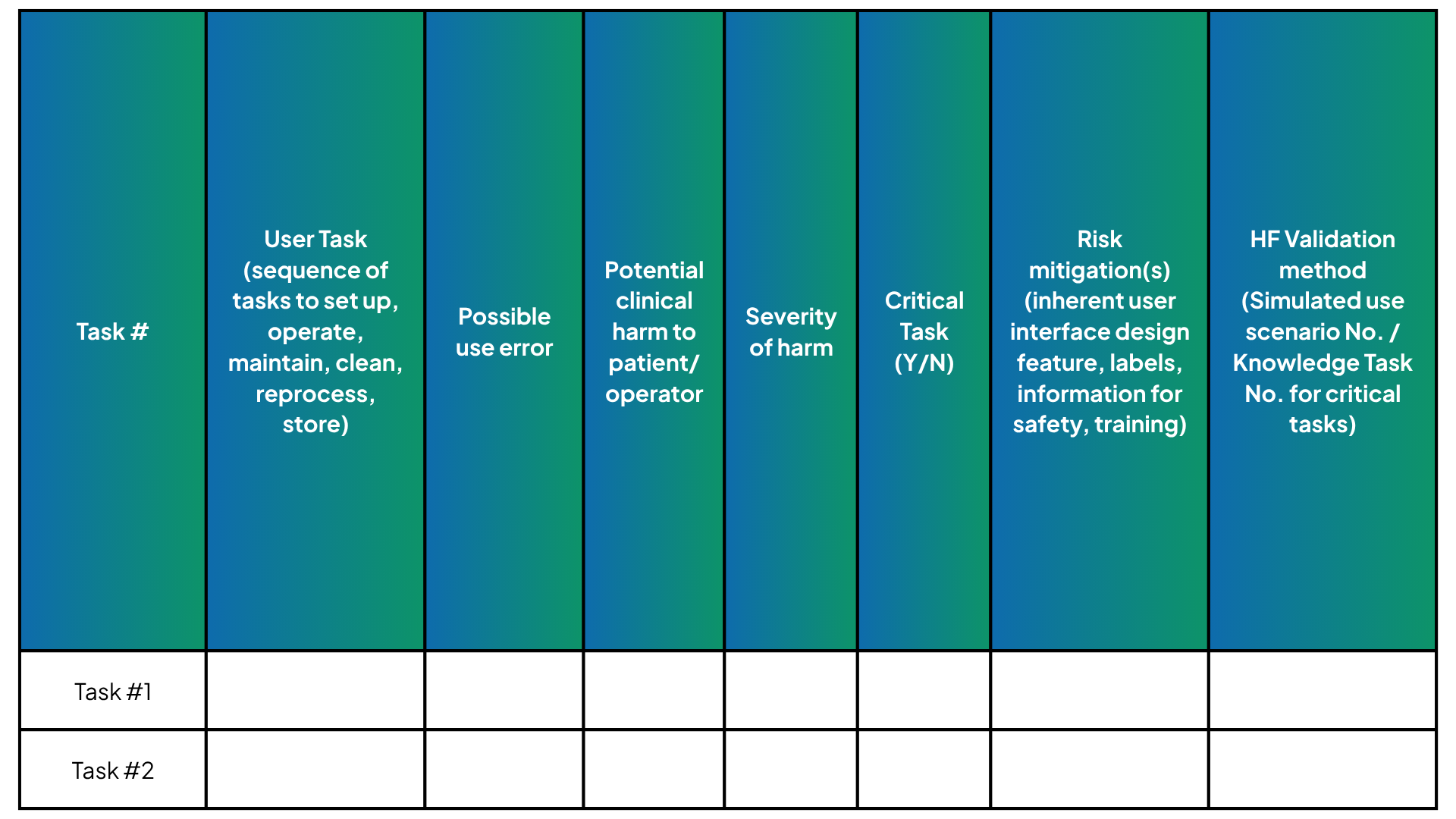
Figura 2: Ejemplo de tarea, control de riesgos, esquema de pruebas
- Informar sobre las tasas de éxito sin establecer la seguridad de uso: Las tasas de éxito numéricas por sí solas son insuficientes. Los fabricantes deben evaluar y analizar los casos de fallo para determinar si incluso errores de uso aislados podrían causar daños graves a los pacientes. El argumento general sobre la seguridad de uso debe basarse en el análisis de errores, no en porcentajes.
- Dependencia excesiva de los datos de preferencias subjetivas: Las valoraciones de facilidad de uso y las preferencias de los usuarios no constituyen prueba de un uso seguro y correcto. Las métricas subjetivas pueden respaldar conclusiones, pero no sustituyen la evaluación objetiva del desempeño en la tarea ni la mitigación de errores. Creer que el dispositivo es fácil de usar no equivale a demostrar un uso correcto ni a presentar un argumento convincente ante los organismos reguladores.
- Representación insuficiente de grupos de usuarios o grupos de usuarios insuficientes: Tanto las directrices de la FDA como la norma IEC 62366-1 recomiendan incluir un mínimo de 15 participantes por grupo de usuarios identificado. Para determinar los grupos de usuarios adecuados, se debe considerar quién realiza las tareas, los perfiles de usuario pertinentes y las diferencias en la formación, la experiencia o las capacidades físicas o cognitivas. Los dispositivos de uso doméstico o los productos combinados para pacientes pueden requerir grupos de usuarios adicionales, como personas con discapacidades visuales o cognitivas. Se recomienda la comunicación previa a la presentación con la FDA para definir la estrategia de pruebas.
- Falta de demora entre la capacitación y las pruebas: Las pruebas de uso simulado deben incluir un intervalo de tiempo entre el entrenamiento y la ejecución de la tarea para tener en cuenta la pérdida de aprendizaje y reflejar las condiciones de uso reales. En algunas solicitudes de productos combinados, la FDA puede exigir un grupo entrenado y otro no entrenado.
- Atribuir los fallos al “error humano” sin análisis: Los resultados de las pruebas sumativas deben analizarse caso por caso, en función de los fallos observados y los patrones de dificultades que presenten los participantes. Atribuir simplemente un fallo a que "el participante lo olvidó" durante la ejecución de una tarea crítica no constituye un argumento sólido para afirmar que la interfaz es segura, ya que esta debe brindar al usuario todo el apoyo posible durante la realización de la tarea. Las entrevistas posteriores a la prueba son fundamentales para comprender el razonamiento del usuario, sus puntos de confusión y los factores causales. Preguntar a los participantes por qué se confundieron, qué les llevó a creer que el dispositivo funcionaba de cierta manera o por qué cometieron un error es absolutamente esencial para el proceso de análisis.
- Fallo en la validación de las instrucciones de uso (IFU) cuando se confían en ellas como control de riesgos principal: Cuando los fabricantes identifican las instrucciones de uso como una medida clave para mitigar riesgos, los organismos reguladores suelen exigir pruebas específicas para validar su eficacia. Las directrices de la FDA destacan que, por lo general, no basta con basarse únicamente en el etiquetado y que, siempre que sea posible, la seguridad debe integrarse en la interfaz de usuario.
Factores humanos y pruebas de usabilidad como ventaja competitiva
Más allá del cumplimiento normativo, las pruebas de factores humanos y de usabilidad ofrecen una ventaja estratégica. Los dispositivos que son intuitivos, eficientes y se adaptan a los flujos de trabajo del mundo real tienen más probabilidades de:
- Ser adoptado por los médicos
- Reducir la carga de capacitación
- Mejorar la satisfacción del usuario
- Minimizar los problemas posteriores a la comercialización
- Fortalecer la confianza en la marca
En un panorama de tecnología médica cada vez más competitivo, la facilidad de uso no es solo un requisito reglamentario, sino un factor diferenciador.
Conclusión
La ingeniería de factores humanos/usabilidad se ha convertido en una parte fundamental del proceso de desarrollo de productos, garantizando la facilidad de uso y la seguridad de los dispositivos médicos. A nivel mundial, los organismos reguladores exigen un proceso sistemático de supervisión y revisión del cumplimiento por parte de los fabricantes de la norma de ingeniería de usabilidad IEC 62366-1. El proceso de revisión previa a la comercialización de la FDA ahora incluye de forma rutinaria el análisis de riesgos relacionados con el usuario y las pruebas de validación de la interfaz de usuario del dispositivo con los usuarios previstos.
Las actividades de factores humanos/experiencia del usuario deben llevarse a cabo en todas las fases del diseño y desarrollo del dispositivo, incluido el análisis preliminar de riesgos relacionados con la tarea y el usuario, el diseño y la evaluación de la interfaz de usuario y las pruebas de validación sumativas finales en un uso simulado.
El proceso de evaluación de factores humanos/experiencia del usuario debe estar alineado con el proceso general de gestión de riesgos del dispositivo. Las decisiones finales sobre la aceptabilidad del riesgo de uso deben basarse en los resultados de pruebas de uso simuladas con usuarios representativos, evaluando la presencia de patrones residuales, fallos y dificultades en tareas con implicaciones de riesgo críticas.
Las pruebas formativas y sumativas de factores humanos desempeñan funciones distintas pero complementarias en el desarrollo de dispositivos médicos. Cuando se aplican estratégicamente, permiten a los fabricantes —independientemente de su tamaño— diseñar dispositivos más seguros, agilizar los trámites regulatorios y reducir el riesgo general de desarrollo.
- Pruebas formativas proporciona la información necesaria para diseñar eficazmente
- Pruebas sumativas proporciona la evidencia necesaria para demostrar seguridad
En conjunto, forman la base de una estrategia sólida y escalable de pruebas de factores humanos y usabilidad que respalda tanto el cumplimiento normativo como el éxito comercial.
Preguntas Frecuentes (FAQ)
¿La usabilidad es diferente de los factores humanos?
El término «ingeniería de usabilidad», a menudo utilizado como sinónimo de «factores humanos», también se centra en crear interfaces de usuario que faciliten el aprendizaje rápido, la satisfacción del usuario y una interacción eficiente. El término «usabilidad» es una cualidad multidimensional que se refiere a la capacidad de una persona para interactuar de forma sencilla y prácticamente sin errores con un sistema o producto.
¿Qué normas o directrices reglamentarias debe tener en cuenta en relación con la usabilidad de los dispositivos médicos?
En Estados Unidos, la FDA reconoce la norma IEC 62366-1, Dispositivos médicos - Parte 1: Aplicación de la ingeniería de usabilidad a los dispositivos médicos, la norma ANSI AAMI HE75, Ingeniería de factores humanos - Diseño de dispositivos médicos y la guía de la FDA, Aplicación de factores humanos e ingeniería de usabilidad a los dispositivos médicos, publicada en febrero de 2016.
La comunidad reguladora internacional ha incorporado la norma IEC 62366-1, Dispositivos médicos – Parte 1: Aplicación de la ingeniería de usabilidad a los dispositivos médicos, como parte del proceso de aprobación fuera de los EE. UU.
¿Son obligatorias las pruebas de factores humanos para todos los dispositivos médicos?
El proceso de revisión previa a la comercialización de la FDA ahora incluye de forma rutinaria el análisis de riesgos relacionados con el usuario y las pruebas de validación de la interfaz de usuario del dispositivo con los usuarios previstos. Esto se observa especialmente en el caso de dispositivos de alto riesgo y software como dispositivo médico (SaMD).
¿Cuántos usuarios se necesitan para las pruebas formativas?
No se requiere un número fijo o mínimo de usuarios para las evaluaciones formativas. Tanto la norma IEC 62366-1 como la guía de la FDA "Aplicación de factores humanos e ingeniería de usabilidad a dispositivos médicos" enfatizan que las pruebas formativas son iterativas y exploratorias, con el objetivo de identificar problemas de usabilidad y riesgos relacionados con el uso, no de validar estadísticamente el rendimiento.
Si bien los organismos reguladores no exigen un tamaño de muestra específico, muchos fabricantes consideran que entre 8 y 10 usuarios representativos pueden revelar el 90 % de los defectos de diseño existentes.
¿En qué se diferencia la evaluación sumativa de otras pruebas de usabilidad?
Existen varias diferencias importantes en la metodología de prueba entre las pruebas de uso simulado sumativas y otras evaluaciones de usabilidad, como las pruebas formativas. La prueba sumativa no tiene como objetivo explorar las características de diseño y debe realizarse sobre el diseño final del dispositivo. Incluye usuarios, entornos y escenarios de uso representativos, y sigue un protocolo estructurado y predefinido que se centra en las tareas críticas y las peores condiciones posibles. Los resultados de la prueba sumativa se incluirán en un informe formal para su presentación ante las autoridades reguladoras.
¿Puede una evaluación formativa rigurosa reducir el riesgo de fracaso en la evaluación sumativa?
Sí. Las pruebas formativas iterativas aumentan significativamente la probabilidad de una validación sumativa exitosa y una revisión regulatoria más fluida.
Recursos y Referencias
Guía de la FDA para la industria y el personal de la Administración de Alimentos y Medicamentos (FDA): Aplicación de factores humanos e ingeniería de usabilidad a dispositivos médicos. Publicada el 3 de febrero de 2016.
ANSI AAMI HE75:2009/(R)2018, Ingeniería de factores humanos – Diseño de dispositivos médicos (reconocido por la FDA n.° 5-57)
IEC 62366-1 Edición 1.1 2020-06 Versión consolidada, Dispositivos médicos – Parte 1: Aplicación de la ingeniería de usabilidad a los dispositivos médicos (reconocido por la FDA n.° 5-129)
ISO 14971 Tercera edición 2019-12, Dispositivos médicos: Aplicación de la gestión de riesgos a los dispositivos médicos (reconocida por la FDA con el número 5-125)
Wiklund, J. Kendler y A. Strochlic, Pruebas de usabilidad de dispositivos médicos. CRC Press-Taylor & Francis Group (2011)
