
On 2026 年 2 月 2 日FDAが長らく待ち望んでいた 品質管理システム規制 (QMSR) が完全に有効になり、従来の品質システム規制 (QSR) に取って代わり、21 CFR Part 820 を正式に整合させた。 ISO 13485:2016これは単なる規制の見直しにとどまらず、医療機器メーカーが今後どのように品質を証明し、リスクを管理し、FDAの査察を受けるかという点において、大きな転換点となるものです。
新しい枠組みはグローバルな期待に沿うように構築されていますが、遵守には戦略的な計画と実質的な運用上の更新の両方が必要です。以下に、変更点とその組織への影響について、明確かつ専門的でありながら分かりやすい概要を示します。
FDA QMSRとは何か、そしてなぜ重要なのか?
その QMSR 米国の医療機器品質要件を現代化する ISO 13485:2016 を参照により組み込む また、特に記録、表示、市販後義務に関する法的要件を維持するために、FDA固有の規定を追加しました。規制の構造は意図的に簡素化されており、QSRの規定的な文言の多くはISOの条項に準拠したものに置き換えられています。
FDAの意図は明確だ。重複する世界的なコンプライアンス負担を軽減し、規制当局間の一貫性を促進し、安全性や有効性の基準を弱めることなく明確性を向上させることである。
主要なFDA QMSR 検査およびコンプライアンスの変更点:何が変わったのか?
FDA QMSR 検査:QSITは廃止されました
新しい QMSR 退職とは 品質システム検査技術(QSIT)2026年2月2日より、すべての機器検査は新しい基準に従うことになります。 コンプライアンスプログラム 7382.850これは、リスクベースでライフサイクルに焦点を当てた検査モデルをサポートするものです。
業界分析は、FDAが示唆してきたことを裏付けている。すなわち、今後の査察は、構造、流れ、文書審査の深さにおいて、これまでとは異なるものになるだろう。製造業者は、従来のQSITに関連付けられていた厳格なサブシステムチェックリストに代わり、より動的でリスク主導型のやり取りを想定すべきである。
リスク管理がFDAを推進する QMSR 検査
新しい検査プログラムは、 QMSR 要件 品質マネジメントシステム(QMS)の6つの領域—経営管理、設計・開発、アウトソーシング・購買を含む—4つの「その他の適用可能なFDA要件」(OAFR)によってサポートされています。 MDR、訂正・撤去、追跡、UDI.
FDAの調査官は現在、 企業のリスク管理ファイルから始めましょうISO 14971に準拠した文書を用いて検査手順を策定します。これにより、適切に維持管理され、継続的に更新されるリスク記録の重要性が高まります。FDAの調査官は、固定されたサブシステムを順にたどるのではなく、この手順に従って、どこに重点を置くべきか、どの程度深く調査すべきか、そして品質システムの問題が組織全体でどのように関連しているかを判断します。
リスクフェーズ:リスク管理ファイルレビュー
検査はリスク管理ファイルのレビューから始まります。FDA QMSRの下では、リスク管理はもはや設計活動に限定されません。調査官はリスク文書を検査全体の出発点およびロードマップとして使用します。このレビューはFDAが以下の点を理解するのに役立ちます。
- デバイスの意図された用途と患者/使用者のリスク
- リスクの特定、評価、管理方法
- リスク管理が設計、製造、サプライヤー、および市販後プロセス全体に一貫して反映されているかどうか
この段階の結果によって、検査時にどの品質マネジメントシステム(QMS)領域がより詳細に調査されるかが決定されます。
QMS領域:コアプロセスの統合レビュー
リスクレビューの後、FDAはCP 7382.850で定義されている6つの品質管理システム(QMS)領域全体にわたるコンプライアンスを評価します。これらの領域は従来のQSITサブシステムに代わるもので、ISO 13485に準拠した構造を反映しています。図に示されているQMS領域は、厳密な順序でレビューされるわけではありません。代わりに、調査官はリスクシグナル、観察された問題、およびプロセス間の関連性に基づいて、これらの領域間を移動します。
- 設計と開発FDAは、設計成果物、検証、妥当性確認、および変更が、特定されたリスクおよび意図された用途に合致しているかどうかを評価します。
- 変更管理設計、プロセス、サプライヤー、またはソフトウェアの変更は、リスクが再評価され、管理策が引き続き有効であることを確認するために見直されます。
- サプライヤーとアウトソーシングサプライヤーの選定、監視、再評価は、リスクベースのアプローチを用いて検討され、必要に応じてサプライヤー監査記録のレビューも含まれます。
- 生産とサービスFDAは、製造およびサービス工程において、仕様およびリスク管理基準を満たす機器が一貫して生産されているかどうかを評価する。
- 測定、分析、改善苦情処理、是正措置・予防措置(CAPA)、傾向分析、データモニタリングを評価し、品質マネジメントシステムが品質上の問題を検知し、対処していることを確認します。
- 経営監視: 経営陣の責任範囲は、包括的な検査対象領域です。FDAは、経営陣が品質マネジメントシステムの有効性、資源配分、および説明責任をどのように確保しているかを審査します。
これらの分野は意図的に相互に関連付けられています。ある分野で懸念事項が特定された場合、FDAは他の分野にも審査範囲を拡大する可能性があります。
OAFR:その他の適用されるFDA要件
QMS領域と並行して、FDAは図に示すその他の適用可能なFDA要件(OAFR)を明確に審査します。
- 医療機器の報告(21 CFR Part 803)
製造業者に対し、医療機器が死亡または重傷の原因となった、あるいはそれに寄与した可能性がある場合、または不具合が再発した場合に重大な危害につながる可能性がある場合に、FDAに報告することを義務付ける。 - 訂正および削除(21 CFR パート 806)
製造業者に対し、健康リスクを低減するため、または規制違反を是正するために、機器の修正または撤去に関して講じた特定の現場措置についてFDAに通知することを義務付ける。 - 追跡(21 CFR Part 821)
製造業者に対し、特定の高リスク医療機器の流通状況と所在を追跡し、必要に応じて迅速な患者への通知や機器の回収を可能にすることを義務付ける。 - 固有機器識別(21 CFR Part 830)
トレーサビリティ、市販後監視、有害事象報告、およびリコール効果を向上させるため、機器に固有の識別子を付けることを義務付ける。
QMSRの下では、これらは正式な検査項目であり、二次的または付随的なレビューではありません。OAFRの不備は、特にリスク管理や市販後のパフォーマンスに関連する場合、検査結果に大きな影響を与える可能性があります。
最終報告:検査概要と所見
検査は、 観察結果の概要これはFDAの評価を反映したものであり、以下の内容を含んでいます。
- リスク管理の有効性
- ライフサイクル全体にわたるQMSの統合
- 経営管理と説明責任
- の遵守 QMSR および該当するFDAの要件
調査結果はFDAの便益・リスクに基づくコンプライアンスフレームワークを用いて評価される。つまり、システム的な問題やリスクに関連する問題は、個別の文書の不備よりも規制上の重要性が高い可能性がある。
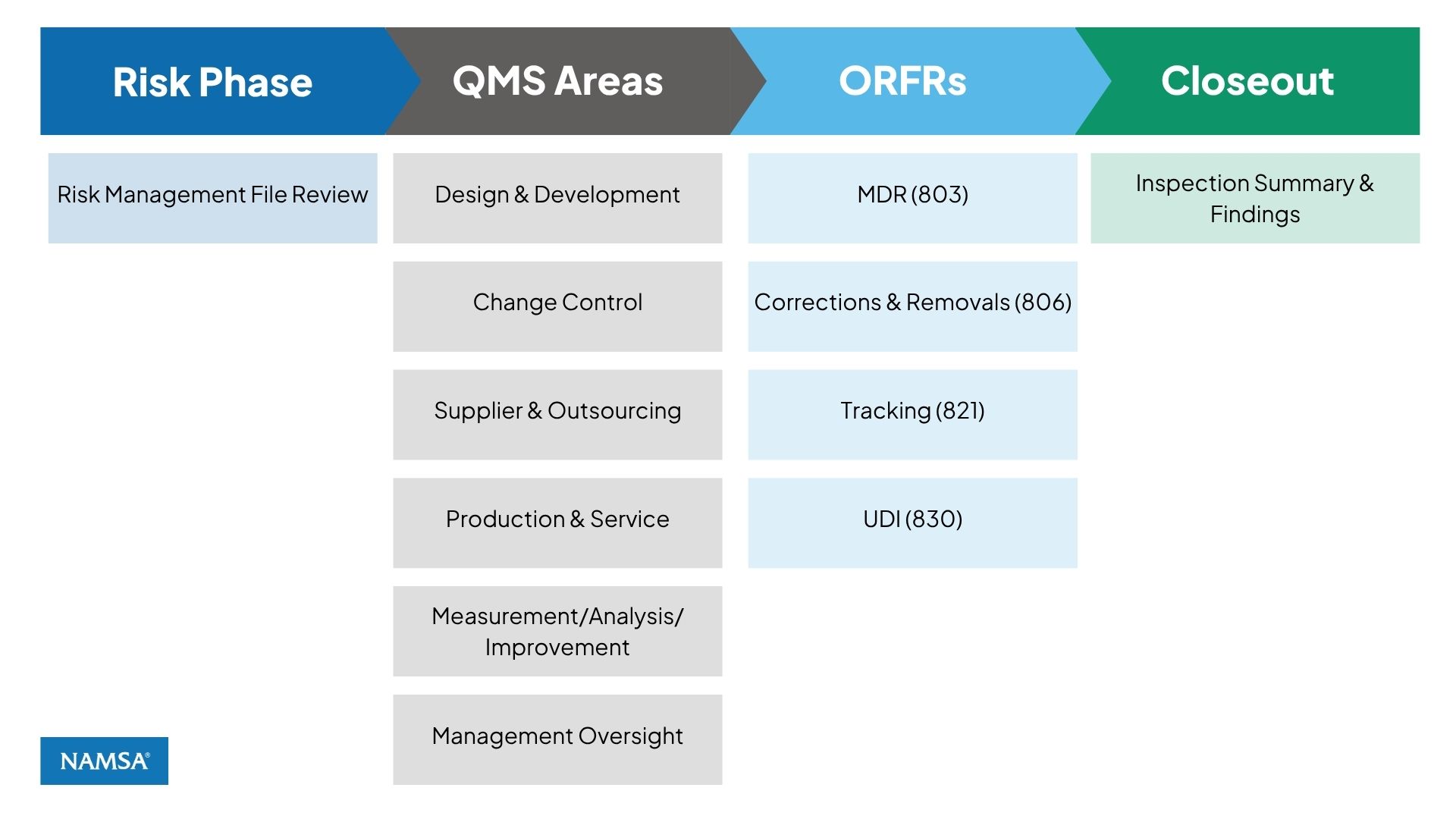
図1FDAの調査官が品質管理システム規則(QMSR)およびコンプライアンスプログラム7382.850に基づいて使用する検査フロー
この流れが重要な理由
この検査の流れはFDAの重要な現実を浮き彫りにしている QMSR 検査:FDAはもはや個々の手順を検査するのではなく、品質システム全体が統合されたリスク主導型の全体としてどのように機能するかを評価している。
製造業者は以下の準備をしておく必要があります。
- リスク管理ファイルから検査を開始する
- リスク、設計、サプライヤー、生産、および市販後データ間の明確な関連性を示す
- 文書化された証拠に基づいて経営判断を裏付ける
- FDAは複数のQMS分野にわたる問題を追跡調査すると予想される。
ISO 13485認証とFDA QMSR:同じものではない
ISO 13485認証を取得している製造業者は有利な立場にあると感じるかもしれないが、 QMSR is ISOの単純な採用。FDAは明確に述べている。 ISO認証は これはFDAの査察に代わるものではなく、また米国特有のすべての要件への準拠を自動的に証明するものでもありません。
分析では、ISO 13485がQMSRの基盤を形成している一方で、用語、記録保持に関する要件、市販後要件など、FDA固有の規定がいくつか存在するため、「ISO準拠」と「QMSR準拠」は同義ではないことが強調されている。
記録的な透明性の向上:内部監査が今や正当な対象となった
おそらく、運用面で最も意義深い変更点は、内部監査、経営陣によるレビュー、およびサプライヤー監査記録をFDAの検査範囲から除外してきた長年の免除規定が廃止されたことでしょう。QMSRの下では、これらの保護措置はなくなります。
さらに、検査ガイドラインの更新により、かつては機密情報または内部向け情報とみなされていたこれらの記録が、現在では定期的なレビューの対象となっていることが確認されました。監査の質、サプライヤーの監督、および経営陣によるレビューの証拠について、より厳格な精査が求められることが予想されます。
連邦官報の技術的修正は、 §820.35(記録の管理) CFRの各パート間で記録管理に関する期待値を統一するために、数十件の相互参照を更新しました。
FDA QMSRに基づく設計および開発
その QMSR 明示的な「設計管理」用語を廃止し、代わりにISO 13485の 設計と開発 フレームワーク。ラベルは異なるものの、検証、妥当性確認、レビュー、転送、変更管理といった根底にある要件は、いずれも同様に厳格である。
ISO 13485では明示的に要求されていませんが、 独立したレビュー担当者 各設計レビュー段階(QSRの慣行と歴史的に関連付けられてきた概念)において、代理店は 製造業者には、適切な専門知識と職務上の責任を持つ人材を関与させるよう引き続き期待している。設計プロセスに対する有意義な監督と客観的な評価を提供できる能力を持つ。FDAは、設計レビューは引き続き実施されなければならないと強調した。 構造化され、文書化され、学際的なISOは独立性の証明方法に関してより柔軟性を認めているとしても、
サプライヤー管理:リスクベースに明確化
ISO 13485のサプライヤー管理は、QMSRを通じて米国の規制に直接組み込まれています。これは、サプライヤーは、以下の基準に基づいて評価、選択、監視、再評価されなければならないことを意味します。 リスクに見合った基準単なる初期資格ではない。
購買管理においては、製造業者はサプライヤーの業績を文書化し、継続的に監視することが義務付けられるようになった。FDA(米国食品医薬品局)もサプライヤーの監査記録を審査するようになる。これは、QSR(クイックサービスレストラン)時代の期待とは大きく異なる点である。
ISOに準拠したガイダンスは、体系的なモニタリング、購買関連文書、およびサプライヤーのパフォーマンス管理に関する明確な証拠の重要性をさらに強調するものです。
FDA QMSRに基づく表示および包装管理
その QMSR 専用のラベル表示および包装要件を組み込んでいます §820.45FDAによるリコールをこれまで引き起こしてきた問題に関する明確性を向上させる。これらの要求はISOの一般的なアプローチを超え、表示および包装プロセスのより厳格な管理と文書化を必要とする。
FDAの規則では、ラベルの混同防止、適切なバージョン管理の確保、ラベルの内容、機器の構成、および固有機器識別番号(UDI)の割り当ての整合性の維持が特に重視されています。さらに、包装の検証では、完全性(および該当する場合は滅菌性)が流通時のストレスに耐え、ラベルに記載された使用期限まで機器の性能を維持することを証明する必要があります。
FDA QMSRの下で変わっていないこと
大規模な構造変化があったにもかかわらず、いくつかの主要な義務は依然として変わっていない。
- その QMSR 依然として適用される 完成品デバイスメーカー安全性と有効性に関する法的要件は維持される。
- 市販後要件には以下が含まれます。 MDR, 修正と削除, UDIこれらは引き続き完全に有効であり、現在では正式な検査項目となっている。
MDSAP:役立つが、代替手段ではない
医療機器単一監査プログラム(MDSAP)は引き続きFDAに受け入れられています 定期的な監視検査の代わりにただし、原因調査、市販前検査、またはコンプライアンス検査の代わりとなるものではありません。MDSAP監査モデル自体は、 QMSR 要件を満たし、グローバルな枠組みをさらに統合する。
FDA QMSRへの製造業者の準備方法
FDAへの準備 QMSR 検査と継続的なコンプライアンスに関して、製造業者は以下を優先すべきである。
1. 品質マニュアルおよびQMSマッピングの更新
手順をISO条項およびQMSR固有の要件、特に§§820.10、820.35、および820.45に合わせる。
2. サプライヤー監視の強化
リスクベースのサプライヤー分類、KPI、監査文書、および変更管理を導入する。
3. 設計記録がISO構造に準拠していることを確認する
従来のDHF/DMR/DHRからISOの設計開発ファイルおよび医療機器ファイルへの明確なマッピングを維持する。
4.リスク主導型検査への準備
リスク管理ファイルから始めて、プロセス全体にわたる問題点を追跡する模擬検査を実施してください。
5. 新しい記録アクセス要件に関する文書とトレーニングの更新
内部監査を監査しましょう。なぜなら、FDAも内部監査を行うからです。
FDA QMSR:より調和のとれた未来へ ― 準備はできていますか?
その QMSR これは、過去30年近くで米国における医療機器品質規制において最も重要な転換点となる。グローバルな慣行に沿ったものであるものの、プロセス、文書化、組織体制の見直しが求められる。効果的に近代化を進める企業は、効率性、グローバルな一貫性、そして検査への対応力を獲得できるだろう。一方、従来のQSR(品質サービス規制)構造やISO認証だけに頼っている企業は、リスクにさらされる可能性がある。
この構造を組織のデバイスポートフォリオに合わせてカスタマイズしたり、構築したりするためのサポートが必要な場合は、 QMSR 移行計画、 NAMSA 喜んでお手伝いいたします。
よくある質問(FAQ)
FDAは QMSR 単にISO 13485を米国法に採用しただけなのか?
いいえ。FDAは QMSR ISO 13485:2016を参照により組み込んでいるだけでなく、ISO認証の範囲を超える、記録、表示、市販後監視、および執行に関するFDA固有の要件も含まれています。
ISO 13485認証は、QMSRに基づくFDAの査察に取って代わるものとなるのでしょうか?
いいえ。ISO 13485:2016はQMSRの基盤を形成していますが、FDAはISO認証がFDAの査察に取って代わるものではなく、記録保持、表示、市販後義務などの米国固有の要件への準拠を自動的に証明するものではないことを明確にしています。「ISO準拠」と「QMSR準拠」は同じではありません。
QMSRの下でリスク管理がより重要になるのはなぜですか?
リスク管理が検査プロセスを推進するようになった。FDAの調査官は、ISO 14971に準拠したリスク文書を用いて検査を開始し、それに基づいてさらに詳細な調査を行うべき箇所を特定する。これにより、品質マネジメントシステム全体にわたって、適切に維持管理され、最新かつ追跡可能なリスク記録の重要性が高まっている。
内部監査や経営陣によるレビューは、FDAの査察対象となったのですか?
はい。FDAのQMSR(品質マネジメントシステム規則)の下では、内部監査、経営陣によるレビュー、およびサプライヤー監査記録はもはや査察免除の対象ではなく、QMSの有効性を評価するために定期的にレビューされる可能性があります。
CAPAが独立した検査サブシステムではなくなった場合でも、依然として重要なのでしょうか?
はい。CAPAはQMSRの下でも依然として重要ですが、FDAは現在、CAPAを独立したプロセスとしてではなく、リスク、苦情、傾向、市販後データによって是正措置がどのように引き起こされるかに焦点を当て、文脈の中で評価しています。
