With the implementation of the MDR, the clinical evaluation requirements have been outlined in Article 61 and Annex XIV (part A), setting forth clearer expectations in comparison to the previous directives. The Clinical Evaluation describes whether the device achieves its intended purpose and is safe for users and patients, and it is subject to rigorous scrutiny by Notified Body (NB) auditors.
Drawing upon our extensive experience and insights from auditor feedback, this article compiles NAMSA’s knowledge since the implementation of the MDR. It provides valuable insights on preparing clinical evaluation documentation that aligns with the MDR requirements.
1. Planning – Is It Too Early to Prepare the Clinical Evaluation?
Despite the importance of the CEP in the clinical evaluation process, auditors have frequently highlighted deficiencies related to its absence or failure to meet MDR requirements. The latter can include a lack of clinical benefits, safety and performance objectives and their related acceptance criteria, unidentified equivalent devices or specific risk determination.
Planning is essential for a successful Clinical Evaluation. By initiating the clinical evaluation process early on, medical device manufacturers can gain insights into the existing gaps in clinical evidence, ensuring a comprehensive and systematic approach to demonstrating the safety and performance of their devices. This planning is done in the CEP, which should include key elements such as the device’s characteristics, methods used to obtain and analyze data, safety and performance objectives and General Safety and Performance Requirements (GSPRs) that require clinical data support. Additionally, the CEP should outline the developmental plan for clinical investigations, covering the device’s life cycle (the Clinical Development Plan).
2. Report Organization – Clear and Coherent
To facilitate the auditors’ evaluation process and minimize potential comments and queries related to not finding the required information, we recommend structuring the CER following the guidelines provided in both the MEDDEV 2.7/1 rev 4 and the MDR. When preparing a CER, it is also worthwhile to review the Medical Devices Coordination Group (MDCG) document 2020-13, which describes the clinical evaluation assessment used by auditors.
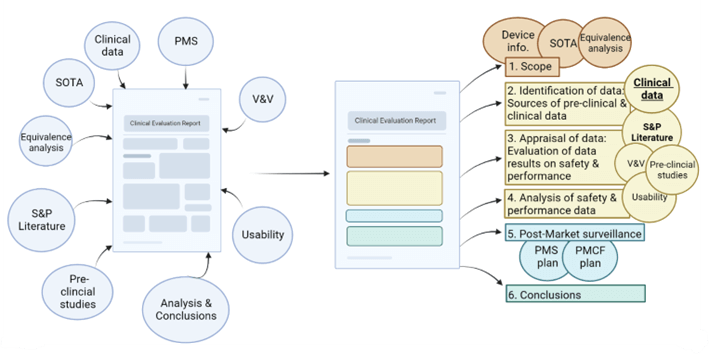
A Clear and Coherent CER: An MDR-compliant template ensures that the data in the CER is organized according to EU guidelines and regulations, making it easier for auditors to find the information. NAMSA has developed a template with input from ex-Notified Body consultants. Our template undergoes continuing improvement as we understand more about how the MDR is being interpreted through interactions with Notified Bodies.
3. State-of-the-Art – Comprehensive and Systematic
When reviewing the state-of-the-art section, two common deficiencies are often highlighted by NB auditors:
a) The state-of-the-art is developed without following a systematic methodology.
Evaluators should use a systematic methodology for identifying and appraising data when developing the state-of-the-art section. It should demonstrate that all standards, guidelines, reviews and articles are selected and appraised unbiasedly.
b) Evaluators often do not use valuable state-of-the-art outcomes when analyzing the device’s data.
We have seen cases in which the state-of-the-art section is only used as an introduction to the medical field, presenting the clinical background of the device under evaluation. However, in addition to establishing the device’s background, the state-of-the-art section should provide valuable information related to the following:
- The evaluation of current treatments and existent associated gaps.
- The development of the clinical claims and objectives to be accomplished by the medical device.
- The identification of similar or equivalent devices.
- The identification of the device’s associated risks (and rates of risk).
- The identification of the device’s associated benefits.
Although the state-of-the-art is a valuable data source, it is sometimes left behind in the benefit-risk analysis section, where the overall data gathered through the clinical evaluation is assessed. When the state-of-the-art has not been included in the benefit risk analysis, NBs may ask for a comparison of the available clinical data to the state-of-the-art.
4. Safety/Performance Claims and Clinical Benefits – Definition and Establishment of the Acceptance Criteria
Auditors have frequently identified deficiencies in the development of claims and acceptance criteria, including reliance on non-published or non-available data.
The articulation of safety and performance claims, as well as clinical benefits, must be clear, unambiguous and supported by clinical data. These claims should be evaluated against pre-established acceptance criteria. Manufacturers should consider all appropriate preclinical and clinical data sources to develop feasible claims and logical acceptance criteria. Its establishment should be defined in the CEP and assessed through the CER, justifying any possible deviation from the acceptance criteria.
5. Equivalence Requirements
The demonstration of equivalence with other devices on the market allows manufacturers to avoid redundant clinical investigations. However, the implementation of the MDR has introduced stricter requirements for comparative evaluations. Notified Bodies pay close attention to whether manufacturers have adequately demonstrated clinical, biological and technical equivalence. To achieve a successful equivalence evaluation, manufacturers must adhere to the MDR and the guidelines provided in MDCG-2020-5.
It is important to note that Class III and implantable devices cannot claim equivalence to competitors’ devices without an agreement to access technical documentation. Manufacturers of lower risk/non-implantable devices can leverage clinical data from competitors’ devices, but claiming equivalence to non-CE marked or non-EU sold products presents additional challenges. For devices classified as Well-Established Technologies (WET), demonstrating similarity with existing devices can help fulfill safety and performance requirements, but scrutiny from Notified Bodies is expected.
6. Additional Considerations
a) Does the CER present sufficient Clinical Evidence?
The thresholds need to be established by the manufacturer, given the risk classification of the device. Appendix III of MDCG 2020-6 can help evaluate the clinical data presented by the device and the hierarchy of clinical evidence to confirm conformity with relevant GSPRs under the MDR.
b) Is the clinical data present in the CER compliant with ISO 14155:2020 Annex A and MDR Annex XV?
If the clinical investigations have been performed outside the EU, the manufacturer needs to assess whether the investigation results may apply to the European population.
c) Does the CER contain opinions?
The evaluation should be objective and transparent. Justifications to explain nuances in the data or deviations from the acceptance criteria, for example, can be included, but they should be referenced and based on reliable scientific information.
d) Are there outstanding gaps in the CER conclusion?
If the gaps are not critical, a Post-Market Clinical Follow-up (PMCF) plan with clear actions to fill these gaps is recommended.
How Can NAMSA Help?
At NAMSA, we understand the complexities and requirements related to the Clinical Evaluation process. Our team of medical writers is comprised of seasoned MDR professionals with extensive experience in guiding medical device manufacturers through the preparation and remediation of technical documentation, including the CEP and CER. Our medical writers partner with our EU Regulatory consultants, many of whom are former Notified Body employees, on CEP and CER development.
In addition to CER medical writing services, at NAMSA, we can provide a wide range of laboratory, quality, regulatory and clinical services to support you in obtaining and maintaining the CE mark for your medical devices.
Contact NAMSA today and let us be your trusted partner on the road to MDR compliance.
Ariadna Navarro
Dr. Ariadna Navarro has a strong scientific background with a PhD in Cardiovascular Sciences and close to ten years of experience in preclinical and clinical research. During her academic career, she collaborated with In Vitro Diagnostic (IVD) manufacturers in the design of strategies and the set up of in vitro techniques to diagnose several cardiovascular and neurological disorders. Dr. Navarro’s medical device industry experience includes working as Clinical Research Scientist and Clinical Study Manager, gaining thorough knowledge in the design, set-up and conduct of clinical investigations according to ICH/GCP guidelines, ISO 14155 and ISO 20916. Ariadna has also developed a strong experience in Regulatory Affairs and Quality Assurance, and she has expert competence on the European regulatory landscape (MDR 2017/745, IVDR 2017/746 and the MEDDEV/MDCG guidance documents). She is a certified ISO 13485 Lead Auditor with experience in setting up medical device quality management system standards aiming to support manufacturers placing and maintain their devices in the market.