Serving Medical Device and IVD Manufacturers in 50+ Countries Worldwide
With experts and labs located throughout the US, Europe, and Japan, NAMSA is ready to support you no matter where you are located.
Contact Us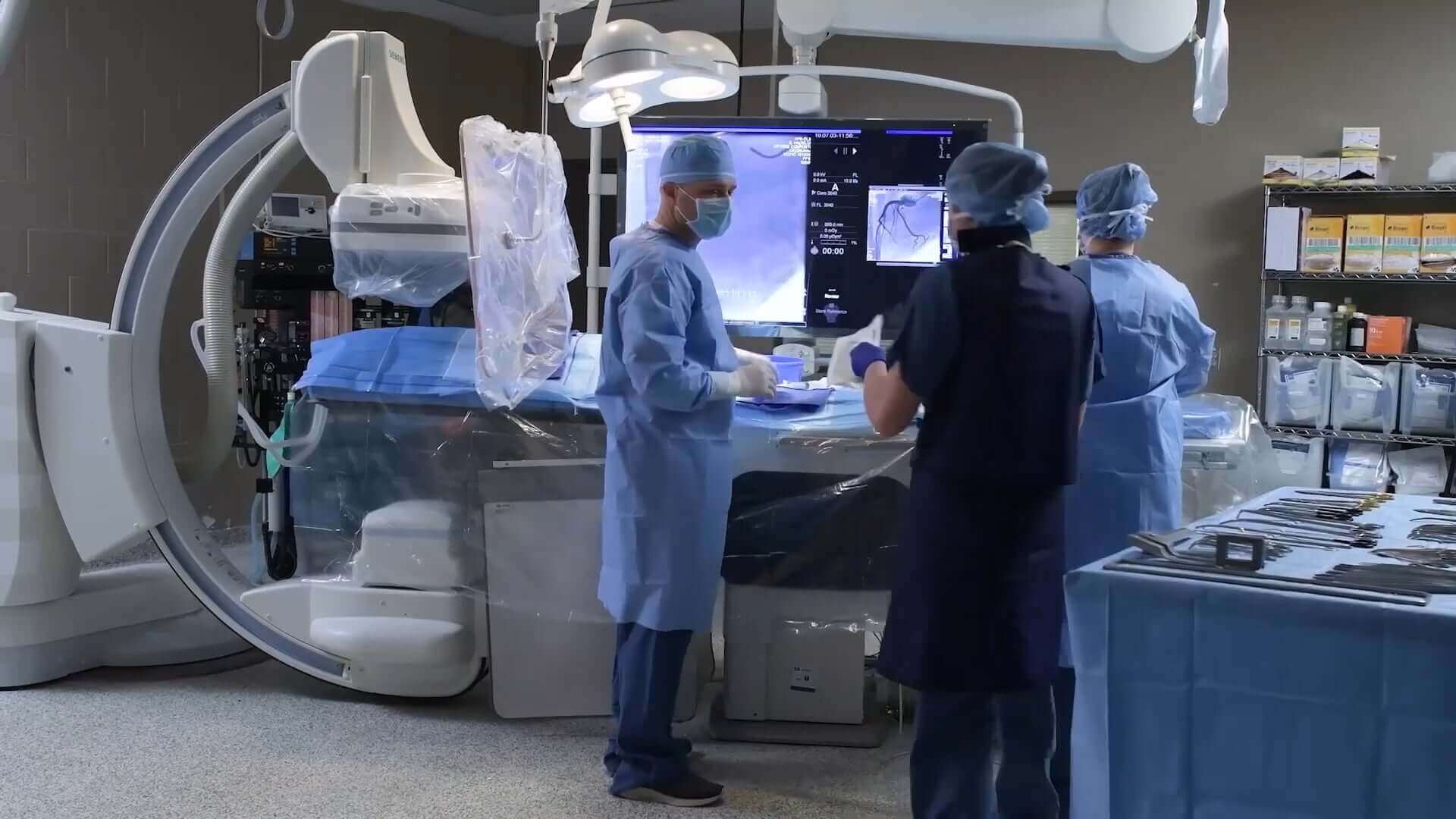
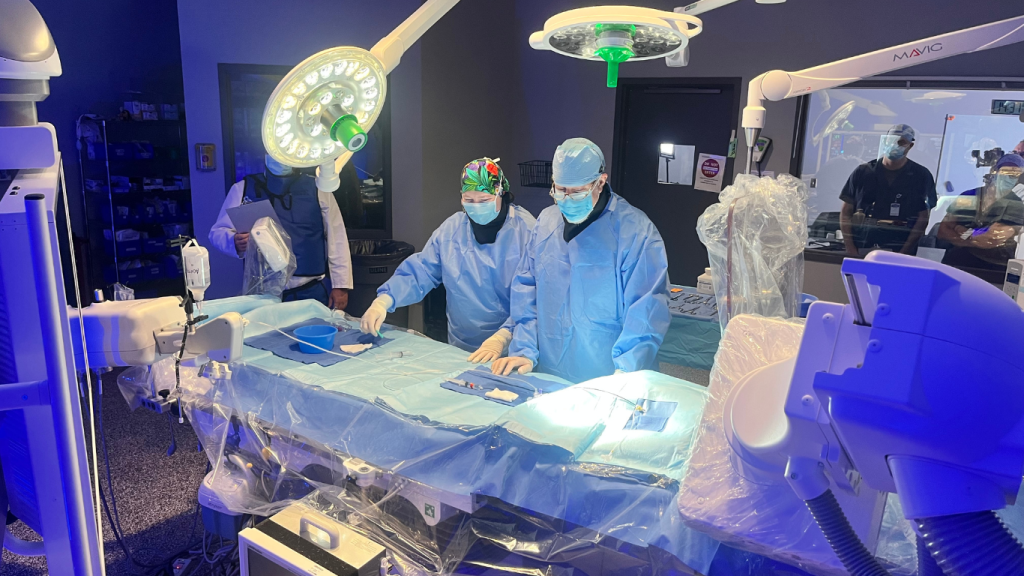
Our team conducts testing, preclinical research, and clinical trials on 100,000+ medical devices each year. From stents to SaMD, we’ve seen it all and we know what the US FDA, EU Notified Bodies, and other global regulators expect from you. Patient and user safety has always been a top NAMSA priority and our team of 1,400+ professionals in North America, Europe, and Asia have the expertise to ensure your medical device is safe, effective, and compliant.
About NAMSA
Year NAMSA Founded to Serve Medical Device Companies
Medical Device or IVD Clinical Projects Each Year
Medical Device and IVD Manufacturers Supported Each Year
Medical Device Tests Conducted Last Year
We provide a wide range of strategic consulting services, from regulatory strategy to post-market support. Our team can assist with medical device biological evaluation plans and reports, CERs, FDA 510(k) and PMA submissions, QMS compliance, PMCF surveys, and market research as a single project or as an extension of your team.
NAMSA has more experience with more devices than any other company focused on medical device research. That equates to unparalleled preclinical laboratory support for sponsors at our GLP-compliant and AAALAC-accredited facilities in the United States and Europe.
Our medical device clinical trial consulting expertise spans every manner of technology, therapy, indication, and geography. This broad range of experience allows us to guide our clients through all phases of clinical research. NAMSA’s global footprint also provides clients direct access to local networks to conduct safe, effective, and efficient clinical trials.
Our state-of-the-art labs conduct over 118,000 biocompatibility, microbiology, sterility, and other tests annually. We take pride in managing all medical device testing services in-house to ensure direct oversight of test articles. NAMSA test results are trusted by the US FDA, European Notified Bodies, and other global regulatory authorities.
MedTech startups often lose valuable time and momentum coordinating multiple vendors across regulatory, preclinical, clinical, and testing activities. While each provider may be capable, the result is often fragmented oversight, unclear timelines, and increased risk — especially in the early stages when decisions matter most.
I worked with [the NAMSA team] to create a biocomp plan for a complicated combination product. They did an excellent job to help us create a solid plan for testing the device that made sense. They did this in a professional manner and listened to my concerns regarding the previous testing we had done. They all delivered a superb product and we couldn’t be happier.
Our deep technical and strategic expertise makes us the preferred choice for medical device and IVD manufacturers in the US, Europe, Japan, and beyond. Meet the people who make NAMSA unique.
Meet the NAMSA Team
With experts and labs located throughout the US, Europe, and Japan, NAMSA is ready to support you no matter where you are located.
Contact Us
Ask a question or request a meeting with a NAMSA expert to discuss your specific project. We will follow up as quickly as possible.