On the heels of the September 26 release of the U.S. Food and Drug Administration’s (FDA), “Digital Health Software Precertification (Pre-Cert) Pilot Program: Tailored Total Product Lifecycle Approaches and Key Findings,” the regulatory authority has released the long-awaited final guidance for “Clinical Decision Support Software” (September 28, 2022). This final guidance replaces the 2017 and 2019 draft guidance documents.
Background
More and more, health care providers are implementing Clinical Decision Support Systems (CDSS) and these solutions are expected to grow in popularity in the near future. Why are CDS systems getting traction? It could certainly be the effect of government initiatives and the COVID-19 Pandemic. But perhaps the primary reason is that they provide clinicians, staff, patients or other individuals with knowledge and person-specific information, intelligently filtered or presented at appropriate times to enhance health and heath care. CDS encompasses a variety of tools to enhance decision-making in the clinical workflow.
‘Clinical Decision Support’ (CDS) is a broad term that encompasses providing “health care professionals (HCPs) and patients with knowledge and person-specific information, intelligently filtered or presented at appropriate times to enhance health and health care,” as mentioned above.
The just-released FDA final guidance:
- Clarifies the scope of the FDA’s CDS software as medical devices intended for HCPs;
- Explains that the FDA’s existing digital health policies continue to apply to software functions that meet the definition of a ‘medical device,’ including those that are intended for use by patients or caregivers; and
- Provides examples of non-device CDS functions and device software functions.
Guidance within 5. Section 3060(a) of the 21st Century Cures Act (Cures Act) amended the Federal Food, Drug and Cosmetic Act (FD&C Act) to add section 520(o) to exclude certain CDS software functions from the definition of a medical device. Software functions that meet all of the following four (4) criterion are not considered medical devices:
Criterion 1: Not intended to acquire, process or analyze a medical image or a signal from an in vitro diagnostic device or a pattern or signal form a signal acquisition system (section 520(o)(1)(E) of FD&C Act).
- The FDA considers software functions that assess or interpret the clinical implications or clinical relevance of a signal, pattern or medical image to be software functions that do not meet Criterion 1 because they acquire, process or analyze. Such devices are subject to FDA regulation and oversite. Note, however, that activity monitors or other signal acquisition systems that measure physiological parameter that are not specifically intended or marketed for a purpose identified in the device definition are not medical devices.
Criterion 2: Intended for the purpose of displaying, analyzing or printing medical information about a patient or other medical information (such as peer-reviewed clinical studies and clinical practice guidelines) (section 520(o) (1)(E)(i) of the FD& C Act).
- Software functions intended to display, analyze or print medical information about a patient or other medical information (e.g. peer-reviewed clinical studies or clinical practice guidelines) meet Criterion 2 and are not medical devices subject to FDA regulation oversight.
Criterion 3: Intended for the purpose of supporting or providing recommendations to a health care professional about prevention, diagnosis or treatment of a disease or condition (section 520(o)(1)(E)(ii) of the FDA& C Act).
- FDA interprets Criterion 3 to refer to software that provides condition-, disease- and/or patient-specific recommendations to an HCP to enhance, inform and/or influence a health care decision (e.g. drug-drug interaction and drug-allergy contraindication notifications to avert adverse drug events), but is not intended to replace or direct an HCP’s judgement and does not include in time-critical decision-making or a specific preventive, diagnostic or treatment output or directive.
- Criterion 3 software functions present recommendations based on an analysis of patient-specific, information to an HCP, who may then incorporate this information into their decision-making about the care of a patient, along with other information and factors of which the HCP is aware. In contrast, software that provides a specific preventive, diagnostic or treatment output or directive that addresses a time-critical decision would be a medical device regulated by FDA and subject to FDA oversight.
- The FDA notes that two aspects of software functionality may affect whether a software function is being used to support or provide recommendations to an HCP: (1) the level of software automation; and (2) the time-critical nature of the HCP’s decision making. Automation bias may occur if software provides an HCP with a single, specific, selected output or solution as opposed to a list of options or complete information for the HCP’s consideration.
Criterion 4: Intended for the purpose of enabling HCPs to independently review the basis for such recommendations that such software presents so that it is not the intent that such health care professional rely primarily on any of such recommendations to make a clinical diagnosis or treatment decision regarding an individual patient (section 520(o)(1)(E) (iii) of the FD&C Act).
- Under Criterion 4, the software function must be intended to enable HCPs to independently review the basis for the recommendation presented by the software so that they do not rely primarily on such recommendations, but rather on their own judgement, to make clinical decisions for individual patients. The FDA explains that regardless of the complexity of the software—and whether or not it is proprietary—the software output or labeling should provide adequate background information in plain language on the input(s), algorithm logic or methods, datasets and validation. Additionally, relevant sources should be identified and available to and understandable by the HCP user.
The graphic below provides a visual overview of the guidance and describes the FDA’s regulatory approach to CDS software functions.
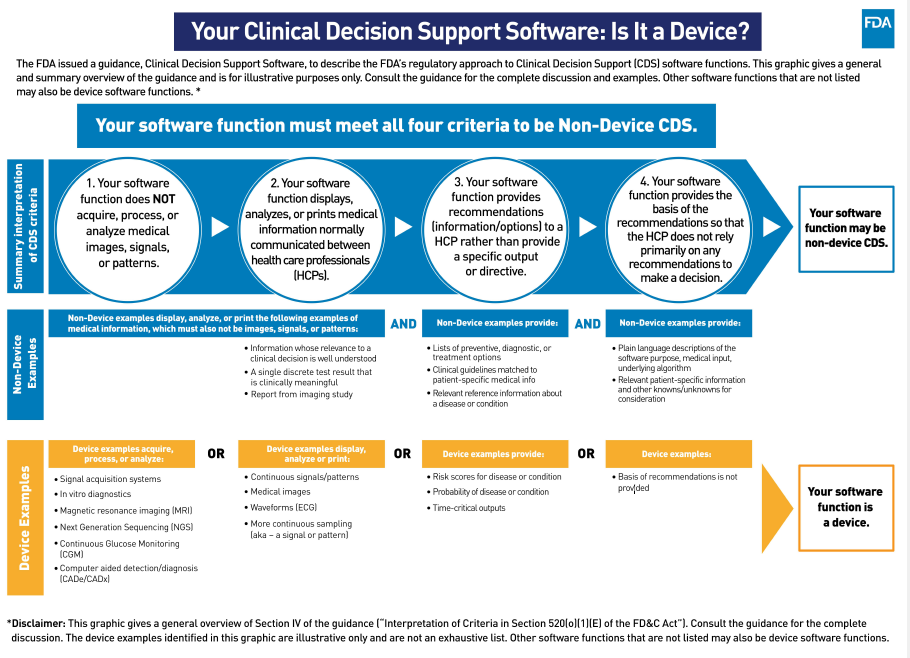
Similar to prior drafts, the purpose of the final guidance is to clarify CDS software functions that:
- Do not meet the definition of a ‘medical device’ as amended by the Cures Act;
- May meet the definition of a device but for which, based on our understanding of the risk of these devices, the FDA does not intend to enforce compliance with applicable device requirements of the FD&C Act (at this time), including—but not limited to—pre-market clearance and pre-market approval requirements; and
- Meet the definition of a device and on which FDA intends to focus its regulatory oversight.
Most Significant Changes
The most significant change from the previous draft guidance to the final guidance is the removal of Section VI: Application of IMDRG Risk Categorization which was replaced by a high level discussion of how risk and a link to the guidance: “Software as a Medical Device”: Possible Framework for Risk Categorization and Corresponding Considerations (imdrf.org). Instead, the FDA’s current thinking regarding risks are discussed under each criterion in more detail, which is helpful.
To be consistent with existing policies, the FDA has updated additional guidance documents at the same time as the release of CDS software guidance. These include:
Policy for Device Software Functions and Mobile Medical Applications
Software as a Medical Device (SAMD): Clinical Evaluation
Medical Device Data Systems, Medical Image Storage Devices, and Medical Image Communications Devices
Medical Device Data Systems, Medical Image Storage Devices, and Medical Image Communications Devices
In addition, the final guidance provides several examples of device and non-device CDS, which should be carefully reviewed by manufacturers when writing an FDA regulatory strategy.
Upcoming FDA Education
The FDA has scheduled a Webinar, “Clinical Decision Support Software Final Guidance” on October 18, 2022 from 1:00 PM -2:15 PM ET. For more information, or to register, visit: https://fda.zoomgov.com/j/1608479998?pwd=SG9ITWNSRGJNMkk0bVZVM29taFphdz09External
Link Disclaimer (Passcode: U6d9c&).
Please Note: Participants who join the webinar using the Zoom webinar link above should use computer audio (listen through computer speakers/speak through computer microphone/headset). The dial-in information provided below is for participants who will be joining the webinar by phone only.
- U.S. Callers, Dial: 833-568-8864 (Toll Free)
- International Callers, Dial: Please check the international numbers available
- Conference ID Number: 160 847 9998
- Passcode: 821003
Following the webinar, a transcript, recording and slides will be made available on CDRH Learn.
How Can NAMSA Help?
NAMSA has experience working with medical device manufacturers who make a wide variety of patient monitoring, disease management, PACS imaging and other software-containing medical devices whose value and effectiveness can be enhanced through mHealth technology. We offer regulatory and quality consulting services for medical device, software as a medical device, IVD and the digital health sector.
We provide consulting services to organizations who design, produce, manufacturer, supply or deploy and use any of the following:
- Software as a Medical Device (SaMD)
- Mobile Medical Apps
- Medical devices of all types with a particular focus on “active” devices and in vitro diagnostic (IVD) medical devices with or without software components or accessories
- Clinical Decision Support and health analytical software
- Software as a Service (SaaS) within healthcare sector
- Artificial Intelligence (AI), deep learning, machine learning, big data algorithms
If you are interested in learning more about these service offerings, please reserve your complimentary consultation today: https://namsa.com/consultants/monica-r-montanez/.
Monica R. Montanez
Monica R. Montanez, MS, RAC, CQA currently serves as NAMSA's Principal Strategy Consultant. Monica has over twenty years’ experience in the medical device industry in Regulatory Affairs and Quality Assurance. Her primary focus is navigating the regulatory pathways for electro-mechanical and software driven medical devices worldwide. She has received clearance of many 510(k)s and approval of new indications for PMA device(s) of which 90% involved software. She has broad regulatory expertise in several areas of digital health, including: Software in a Medical Device (SiMD), Software as a Medical Device (SaMD), mobile medical apps, clinical decision support software, telehealth, artificial intelligence, machine learning, interoperability, cybersecurity and human factors engineering, including wireless medical devices -radio frequency (RF), electromagnetic compatibility (EMC) and electromagnetic interference (EMI). While in industry, she assisted in the development of FDA 510(k) guidance and FDA Software guidance directly with FDA. Monica holds a Masters of Science (MS) degree in Regulatory Science (RS) from the University of Southern California (USC) School of Pharmacy. Currently. she holds Regulatory Affairs Certification (RAC) from the Regulatory Affairs Professionals Society (RAPS) and Certified Quality Auditor (CQA) from the American Society for Quality (ASQ).