As of 26 May 2022, the IVDR as amended by Regulation (EU) 2022/112 has come into force and replaced the IVD Directive 98/79/EC. As a result of this transition, pending drug clinical trials that enroll diagnostic tests that fall under this new IVDR regulation, may be affected. In May 2022, a Medical Device Coordination Group (MDCG) document was published covering important information about the implementation of EU Regulation 2017/746 and how guidance relates to Clinical Trial Regulation (CTR).
The need for interoperability of databases for clinical trials with medicinal products and medical devices is explained within the new regulations, however, there are no further details on the interface of the IVDR and CTR in the legislation (e.g. the requirements for assays used in the context of clinical trials). There is no dedicated procedure foreseen in the mentioned regulations and therefore, no harmonized procedure currently in place.
Assays used in clinical trials may range from CE-marked IVDs to trial- or medicinal product-specific assays that are not always meant to be developed as IVDs. In order to assure that conformity is maintained on both the drug and device sides, MDCG—with the Clinical Trials Expert Group (CTG)—have published a Q&A document to clarify requirements for these assays.
Much focus has been placed in the document on the concept of the “medical purpose of an assay in a clinical trial.” This is achieved by providing clarification on the regulatory status of assays performed on human samples used in the context of clinical trials, as well as on the regulatory expectations toward the clinical trial Sponsors. The overall aim of the document is to support the conduct of clinical trials using diagnostic assays, including combined trials for the development of companion diagnostics (CDx).
It is stated that non-interventional studies, defined as clinical studies other than a clinical trial in Article 2 (4) of the CTR are out of scope of both the CTR (as per Article 1) and this MDCG document. However, IVDs used in non-interventional clinical studies must adhere to the IVDR. This makes sense, since non-interventional studies used to demonstrate the performance and effectiveness of an IVD will now fall under the scope of the IVDR. However, these studies will not fall under the scope of a drug clinical trial as these are interventional by default; this document deals only with the use of (IVD) assays in support of clinical trials.
The challenge is what happens when an existing assay that is already enrolled in a clinical trial falls into the definition of an IVD, per Article 2 IVDR after 26 May 2022.
- Is it now that the IVD device has not been placed on the market or made available under the IVDD after this date?
- Would the product now need to undergo a conformity assessment per IVDR, requiring a formal application to a Notified Body?
- Or, what happens if an IVD has been used in a clinical trial but outside the scope of its intended purpose by the clinical trial Sponsor?
- Can the laboratory results obtained with the assay still be used to support the conformity of medicinal products (especially, when a medicinal product is investigated per CTR, simultaneously with and IVD [Clinical Performance Studies])?
These can become tough questions.
Current State of Play of the IVDR: Potential Affects to Clinical Trials and IVD Manufacturers
The IVDR explains rules concerning the “placing on the market,” “making available on the market” or “putting into service” of IVDs and accessories for an IVD. It further outlines the requirements for performance studies concerning IVDs and accessories for an IVD conducted in the European Union (EU).
In the EU, the following scenarios are possible when using assays in clinical trials:
- The assay used in the clinical trial is an IVD, has an intended purpose meeting the definition of an IVD medical device, in accordance to Article 2, IVDR. The assay has been placed on the market and made available under the IVDD in the EU.
- The assay used in the clinical trial is not qualified as an IVD, has not been placed on the market and/or made available under the IVDD in the EU; but has an intended purpose, making it meeting the definition of an IVD medical device, in accordance to Article 2 of the IVDR.
- The assay used in the clinical trial is intended for research use only purposes, however, has been applied for (and approved to) be used in a clinical trial. The assay does not meet the definition of an IVD medical device, in accordance to Article 2 of the IVDR.
- The assays used in the clinical trial qualify as an in-house developed IVD, in accordance to the definitions in the IVDR.
- The assays used in the clinical trial are a device for performance study in accordance to the definitions in the IVDR.
The MDCG document breaks it all down in a series of questions and answers that address these different scenarios.
The most important takeaways from the MDCG are the following:
- The intended purpose defines whether an assay would fall under the regime of the IVDR or not, irrespective how the assay is regulatory qualified, originally. Not all assays that are used in a clinical trial are subjected to the IVDR, but those that fulfil the definition of an IVD (per Article 2, IVDR) are. Such assays may have a medical purpose within the clinical trial (e.g. when they guide medical management decisions or follow-up).
- An assay is considered an IVD if the manufacturer assigns an intended purpose that fulfils the definition of an IVD according to IVDR Article 2. Where a clinical trial Sponsor assigns a medical purpose to an assay in the context of the clinical trial in a way that the assay fulfils the definition of an IVD according to IVD Regulation 2017/746 Article 2, the clinical trial Sponsor may assume the role of a manufacturer under the IVDR. In this role, it is up to the clinical trial Sponsor to determine the regulatory status of the assay based on the planned use in the clinical trial. When assuming the role of a “manufacturer” this may also mean it may place the device on the market for a different intended purpose or indication, for performance study or investigational use (it is not acceptable to use an investigational IVD without evaluating its performance).
- IMAExamples of assays used in clinical trials that are IVDs, as mentioned under Q06 of the MDCG document, include assays providing information for clinical trial-related medical management decisions (typically to select patients for enrolment in the trial, assign patients to a treatment arm, etc.) and/or may be used to guide follow up measures during and beyond the clinical trial. This would, for example, not be the case, in settings where all trial participants are tested irrespective of treatment arm or medical management and the analysis of impact is conducted retrospectively and where medical management is not impacted by assay results. See also the figure below that resembles a medical purpose (likely to be considered IVDs) in blue and not a medical purpose (no IVDs) in pink.
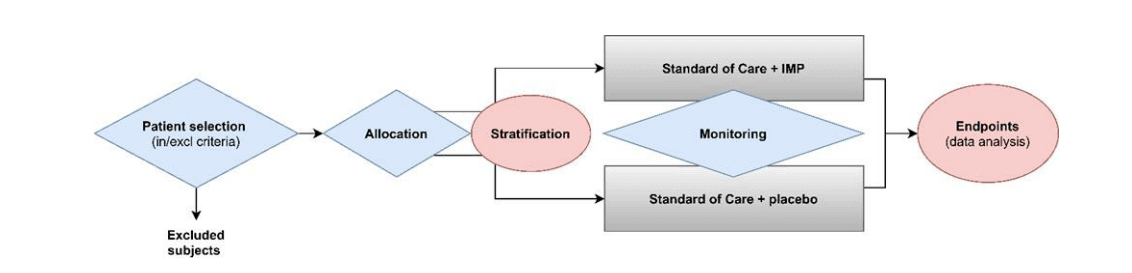
Image Source: MDCG 2022-10
- Non CE-marked IVDs typically used in a clinical trial for a medical purpose in accordance to the IVDR can either be a device for performance study or an in-house IVD.
- Assays that are intended for research use only and are assigned a medical purpose by the clinical trial Sponsor in the clinical trial protocol in a way that the assay fulfils the definition of an IVD according to IVDR Article 2, will become an IVD and cannot be considered a research use only assay. Also, it is interesting to note that, in this situation, the Sponsor will be considered as the ‘manufacturer’ and will have to assume this role under IVDR.
- In-house devices can be used as long as the provisions of the IVDR and relevant national legislations are satisfied, which notably include, that the device must be manufactured and used within a health institution established in the EU and the device is not transferred to another legal entity. However, samples can travel and be analysed by means of in-house
devices based in a different location than the trial site facilitating a core lab facility approach. - Only IVDs specifically required by the protocol to be used to achieve the objectives of the clinical trial, are to be mentioned in the cover letter toward the competent authorities. The information provided should permit identification of the IVD being used and whether it is CE-marked for the planned use. It is the responsibility of the clinical trial Sponsor to determine whether the device is used in the clinical trial and is in line with the manufacturer’s stated intended purpose. In case the device is used outside the intended purpose, the clinical trial Sponsor is then assumed to take responsibility of a manufacturer for that part that is outside the intended purpose, similar to the situation with research use only device. Use of an IVD in a clinical trial may meet the definition of a device for performance study—in this case, when this can be contractually arranged with the original IVD manufacturer.
Important Considerations for Guidance Implementation
In summary, the new MDCG document provides much clarity on the use of assays in clinical trials and the ramifications of the IVDR after the Date of Application (26 May 2022). It must be noted that, these ramifications may be limited, depending on whether an assay (IVD) has already been placed on the EU market before that date, and whether the device is used as intended by the manufacturer during the clinical trial by the clinical trial sponsor.
Potential challenges may arise when assays that are intended for research use only are attributed a different (medical) intended purpose or when a (CE-marked) IVD that is approved for a particular intended purpose is now used for a different intended purpose in a clinical trial. Understandably, the role of an assay varies within a clinical trial and some parts of the clinical trial where assays are used (such as stratification and end-point analysis) will not fall in the category of a medical intended purpose. Nevertheless, a proper assessment on pending clinical trial applications of the impacts of the new regulatory requirements with the MDCG 2022-10 in mind are certainly recommended, especially when clinical trial applications need to be sent to the competent authorities.
Some important recommendations of the MDCG 2022-10 for manufacturers and clinical trial Sponsors to be noted are:
- Read the MDCG 2022-10 alongside what is already mentioned in the IVDR, especially after its amendment through Regulation (EU) 2022/112; since the document will help you strategize your regulatory IVDR roll-out plan.
- Review and revisit the clinical protocols for control of assays in clinical trials; the authorities will expect you to do this.
- Use this information in your advantage to further improve your understanding of intended purpose and device related aspects in clinical trials.
How Can NAMSA Help?
At NAMSA, our European regulatory team can help guide you through this complex regulatory environment and help determine the correct regulatory pathway for your IVD product. We not only understand how to accurately interpret the complicated regulatory challenges that IVD manufacturers sometimes face, but we also help you simplify the development and implementation of effective development strategies. Whether supporting IVD regulatory assessments and submissions, developing IVDR compliant technical files, designing and managing clinical trials or building ISO 13485:2016 and 21 CFR part 820 compliant quality systems… we’ve got you covered.
To learn about NAMSA’s full suite of IVD services and solutions, including IVDR compliance planning, please visit: https://namsa.com/services/ivd/. Or, if you’re ready to set up a complimentary consultation, get in touch with one of our IVD experts here: https://namsa.com/namsa-expertise/subject-matter-experts.
Alex Laan
Alex Laan has been in the IVD and Medical Device industry for a total of 20 years with 12 years spent working for a respectable EU Notified Body. Alex has held the position of Principal Certification Manager at DEKRA Certification BV, a Notified Body for IVDs and Medical Devices located in Arnhem, The Netherlands. Mr. Laan also worked for KEMA Quality, beginning in 2006, where he was qualified for Lead Assessor for Medical Devices and IVD devices, Device Specialist reviewer in IVDs and Medical Devices, including drug-device combinations. In these roles, Alex managed projects with large multinationals (e.g., Boston Scientific), as well as small start-up companies. Alex has been an active member of IVD working group within EU Notified Bodies and TEAM-NB and acting as direct contact for EU competent authorities and EMA on implementation of the EU IVDR (2017/746) and EU MDR (2017/745).