
On February 2, 2026, the FDA’s long‑anticipated Quality Management System Regulation (QMSR) became fully effective, replacing the legacy Quality System Regulation (QSR) and formally aligning 21 CFR Part 820 with ISO 13485:2016. This is more than a regulatory refresh—it represents a significant shift in how medical device manufacturers will demonstrate quality, manage risk, and undergo FDA inspection going forward.
While the new framework is built to harmonize with global expectations, compliance will require both strategic planning and meaningful operational updates. Below is a clear, professional, but approachable overview of what is changing—and what it means for your organization.
What Is the FDA QMSR, and Why Does It Matter?
The QMSR modernizes U.S. medical device quality requirements by incorporating ISO 13485:2016 by reference and adding targeted FDA‑specific provisions to preserve statutory requirements—particularly around records, labeling, and post‑market obligations. The regulation’s structure is now intentionally streamlined, with much of the prescriptive QSR text replaced by alignment with ISO clauses.
The FDA’s intent is clear: reduce redundant global compliance burdens, promote consistency across regulatory authorities, and improve clarity without weakening safety or effectiveness standards.
Key FDA QMSR Inspection and Compliance Changes: What’s Different?
FDA QMSR Inspections: QSIT Is Retired
One of the most consequential changes with the new QMSR is the retirement of the Quality System Inspection Technique (QSIT). Effective February 2, 2026, all device inspections will follow the new Compliance Program 7382.850, which supports a risk‑based, lifecycle‑focused inspection model.
Industry analyses confirm what the FDA has signaled: future inspections will look different in structure, flow, and depth of document review. Manufacturers should expect more dynamic, risk‑driven interactions in place of rigid subsystem checklists previously associated with QSIT.
Risk Management Drives FDA QMSR Inspections
The new inspection program organizes QMSR requirements into six Quality Management System (QMS) Areas—including Management Oversight, Design & Development, and Outsourcing & Purchasing—supported by four “Other Applicable FDA Requirements” (OAFRs): MDR, Corrections & Removals, Tracking, and UDI.
FDA investigators are now expected to begin with a company’s risk management file, using ISO 14971‑aligned documentation to shape the inspection pathway. This expands the importance of well‑maintained, continuously updated risk records. Rather than moving through fixed subsystems, FDA investigators follow this sequence to determine where to focus, how deeply to review, and how quality system issues connect across the organization.
Risk Phase: Risk Management File Review
The inspection begins with a review of the risk management file. Under FDA QMSR, risk management is no longer confined to design activities. Investigators use risk documentation as the starting point and roadmap for the entire inspection. This review helps the FDA understand:
- The device’s intended use and patient/user risks
- How risks were identified, evaluated, and controlled
- Whether risk controls are reflected consistently across design, production, suppliers, and postmarket processes
The outcome of this phase determines which QMS areas receive deeper scrutiny during the inspection.
QMS Areas: Integrated Review of Core Processes
Following the risk review, the FDA evaluates compliance across the six Quality Management System (QMS) Areas defined in CP 7382.850. These areas replace the legacy QSIT subsystems and reflect an ISO 13485‑aligned structure. The QMS Areas shown in the diagram are not reviewed in a rigid order. Instead, investigators move between them based on risk signals, observed issues, and process linkages:
- Design and Development: The FDA assesses whether design outputs, verification, validation, and changes align with identified risks and intended use.
- Change Control: Changes to design, processes, suppliers, or software are reviewed to ensure risks are reassessed and controls remain effective.
- Supplier & Outsourcing: Supplier selection, monitoring, and reevaluation are examined using a risk‑based approach, including review of supplier audit records, when relevant.
- Production & Service: The FDA evaluates whether manufacturing and service processes consistently produce devices that meet specifications and risk controls.
- Measurement, Analysis, and Improvement: Complaint handling, CAPA, trend analysis, and data monitoring are assessed to confirm the QMS detects and addresses quality issues.
- Management Oversight: Management responsibility is a full inspection area. The FDA reviews how leadership ensures QMS effectiveness, resource allocation, and accountability.
These areas are intentionally interconnected. A concern identified in one area may lead the FDA to expand review into others.
OAFRs: Other Applicable FDA Requirements
In parallel with the QMS Areas, the FDA explicitly reviews the Other Applicable FDA Requirements (OAFRs) shown in the diagram:
- Medical Device Reporting (21 CFR Part 803)
Requires manufacturers to report to the FDA when a device may have caused or contributed to a death or serious injury, or when a malfunction could lead to serious harm if it recurs. - Corrections and Removals (21 CFR Part 806)
Requires manufacturers to notify the FDA of certain field actions taken to correct or remove devices to reduce a risk to health or remedy a regulatory violation. - Tracking (21 CFR Part 821)
Requires manufacturers to track the distribution and location of certain high‑risk devices to enable rapid patient notification or device recall if needed. - Unique Device Identification (21 CFR Part 830)
Requires devices to carry a unique identifier to improve traceability, postmarket surveillance, adverse event reporting, and recall effectiveness.
Under QMSR, these are formal inspection components, not secondary or satellite reviews. Deficiencies in OAFRs can significantly influence inspection outcomes, particularly when linked to risk management or postmarket performance.
Closeout: Inspection Summary and Findings
The inspection concludes with a summary of observations and findings, reflecting the FDA’s assessment of:
- Risk management effectiveness
- QMS integration across lifecycle stages
- Management oversight and accountability
- Compliance with QMSR and applicable FDA requirements
Findings are evaluated using the FDA’s benefit‑risk‑informed compliance framework, meaning systemic or risk‑related issues may carry greater regulatory significance than isolated documentation gaps.
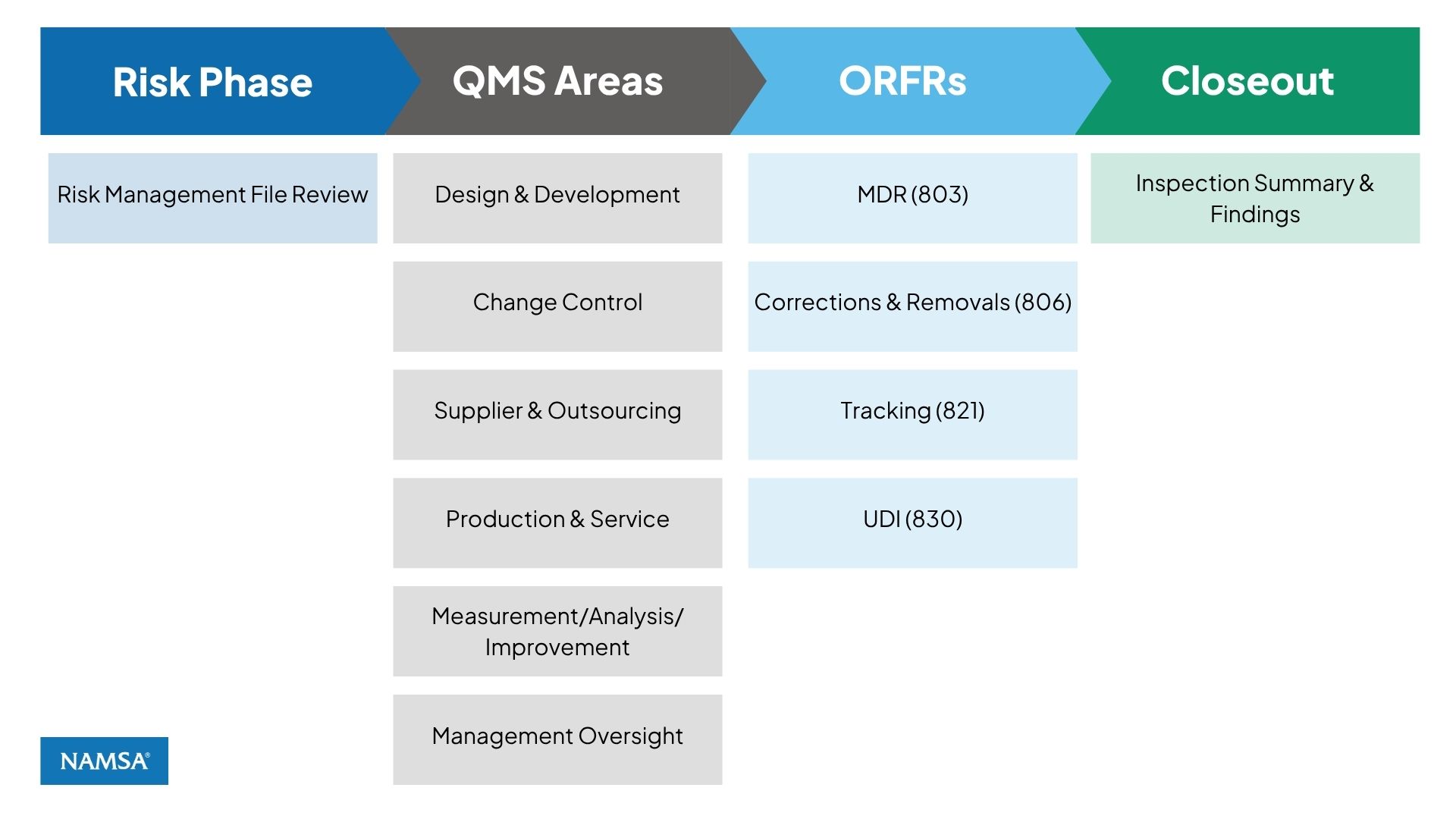
Figure 1: Inspection flow used by FDA investigators under the Quality Management System Regulation (QMSR) and Compliance Program 7382.850
Why This Flow Matters
This inspection flow highlights a key reality of FDA QMSR inspections: the FDA is no longer inspecting isolated procedures — it is evaluating how the entire quality system functions as an integrated, risk‑driven whole.
Manufacturers should be prepared to:
- Start inspections with their risk management file
- Demonstrate clear linkages between risk, design, suppliers, production, and postmarket data
- Support management decisions with documented evidence
- Expect the FDA to follow issues across multiple QMS areas
ISO 13485 Certification and FDA QMSR: Not the Same Thing
Manufacturers with ISO 13485 certification may feel well positioned, but the QMSR is not a simple adoption of ISO. The FDA has explicitly stated that ISO certification will not replace an FDA inspection, nor does it automatically demonstrate compliance with all U.S.‑specific requirements.
Analyses emphasize that while ISO 13485 forms the backbone of the QMSR, several FDA‑specific provisions—including terminology, record‑keeping expectations, and post‑market requirements—mean that “ISO‑compliant” and “QMSR‑compliant” are not synonymous.
Record Transparency Increases: Internal Audits Are Now Fair Game
Perhaps the most operationally meaningful update is the elimination of the long‑standing exemption that kept internal audits, management reviews, and supplier audit records out of the FDA’s inspection scope. Under the QMSR, those protections are gone.
Furthermore, inspection guidance updates confirm that these records—once considered confidential or internal‑facing—are now part of routine review. Expect deeper scrutiny of audit quality, supplier oversight, and management review evidence.
The Federal Register’s technical amendments reinforce the centrality of §820.35 (Control of Records) by updating dozens of cross‑references to align recordkeeping expectations across CFR Parts.
Design and Development Under FDA QMSR
The QMSR retires the explicit “Design Controls” terminology, instead relying on ISO 13485’s Design and Development framework. While the labels are different, the underlying requirements—verification, validation, review, transfer, and change management—remain equally rigorous.
While ISO 13485 does not explicitly require an independent reviewer at each design review stage (a concept historically associated with QSR practices), the agency still expects manufacturers to involve personnel with appropriate expertise and functional responsibility, capable of providing meaningful oversight and objective evaluation of the design process. The FDA emphasized that design reviews must remain structured, documented, and multidisciplinary, even if ISO allows more flexibility in how independence is demonstrated.
Supplier Management: Now Explicitly Risk‑Based
ISO 13485’s supplier controls are now directly embedded into U.S. regulation via QMSR. This means suppliers must be evaluated, selected, monitored, and reevaluated based on risk‑proportionate criteria, not simply initial qualification.
Purchasing controls now require manufacturers to maintain documented, ongoing oversight of supplier performance. The FDA will also review supplier audit records—another major shift from QSR-era expectations.
ISO‑aligned guidance further reinforces the importance of structured monitoring, purchasing documentation, and clear evidence of supplier performance management.
Labeling and Packaging Controls Under FDA QMSR
The QMSR incorporates dedicated labeling and packaging requirements under §820.45, improving clarity around topics that have historically driven FDA recalls. These expectations go beyond ISO’s generalized approach and require tighter control and documentation of labeling and packaging processes.
The FDA’s rule specifically emphasizes preventing labeling mix‑ups, ensuring correct version control, and maintaining alignment between labeling content, device configuration, and Unique Device Identification (UDI) assignment. Additionally, packaging validations must provide evidence that integrity (and sterility, when applicable), withstands distribution stresses, and preserves device performance through the labeled shelf life.
What Has Not Changed Under FDA QMSR
Despite major structural changes, several core obligations remain unchanged:
- The QMSR still applies to finished device manufacturers, with statutory expectations for safety and effectiveness preserved.
- Post‑market requirements, including MDR, Corrections & Removals, and UDI, remain fully in effect and are now formal inspection components.
MDSAP: Helpful but Not a Substitute
The Medical Device Single Audit Program (MDSAP) continues to be accepted by the FDA in lieu of routine surveillance inspections, but not in place of for‑cause, premarket, or compliance inspections. The MDSAP audit model itself has been updated to reflect QMSR requirements, further integrating global frameworks.
How Manufacturers Should Prepare for FDA QMSR
To prepare for FDA QMSR inspections and ongoing compliance, manufacturers should prioritize:
1. Updating the Quality Manual and QMS Mapping
Align procedures with ISO clauses and QMSR‑specific requirements—especially §§ 820.10, 820.35, and 820.45.
2. Strengthening Supplier Oversight
Implement risk‑based supplier classification, KPIs, audit documentation, and change controls.
3. Ensuring Design Records Meet ISO Structure
Maintain clear mapping from legacy DHF/DMR/DHR to ISO’s Design & Development File and Medical Device File.
4. Preparing for Risk‑Driven Inspections
Conduct mock inspections that begin with your risk management file and follow issues across processes.
5. Updating Documentation and Training for New Record Access Expectations
Audit your internal audits—because the FDA will too.
FDA QMSR: A More Harmonized Future—If You’re Ready
The QMSR marks the most significant shift in U.S. device quality regulation in nearly 30 years. While aligned with global practices, it demands a fresh look at processes, documentation, and organizational readiness. Companies that modernize effectively will gain efficiency, global consistency, and inspection‑readiness. Those that rely solely on legacy QSR structures or ISO certifications may find themselves exposed.
If you’d like help tailoring this structure to your organization’s device portfolio or building a QMSR transition plan, NAMSA is happy to assist.
Frequently Asked Questions (FAQs)
Is the FDA QMSR simply ISO 13485 adopted into U.S. regulation?
No. While the FDA QMSR incorporates ISO 13485:2016 by reference, it also includes FDA‑specific requirements related to records, labeling, postmarket surveillance, and enforcement that go beyond ISO certification.
Will ISO 13485 certification replace an FDA inspection under QMSR?
No. While ISO 13485:2016 forms the backbone of the QMSR, the FDA has made it clear that ISO certification does not replace an FDA inspection and does not automatically demonstrate compliance with U.S.‑specific requirements, including recordkeeping, labeling, and postmarket obligations. “ISO‑compliant” and “QMSR‑compliant” are not the same.
Why is risk management more important under the QMSR?
Risk management now drives the inspection pathway. FDA investigators begin inspections with ISO 14971‑aligned risk documentation, using it to determine where to probe deeper. This elevates the importance of well‑maintained, current, and traceable risk records across the QMS.
Are internal audits and management reviews now subject to FDA inspection?
Yes. Under FDA QMSR, internal audits, management reviews, and supplier audit records are no longer inspection‑exempt and may be routinely reviewed to assess QMS effectiveness.
Does CAPA still matter if it is no longer a standalone inspection subsystem?
Yes. CAPA remains critical under QMSR, but the FDA now evaluates it in context, focusing on how corrective actions are triggered by risk, complaints, trends, and postmarket data rather than as an isolated process.
