
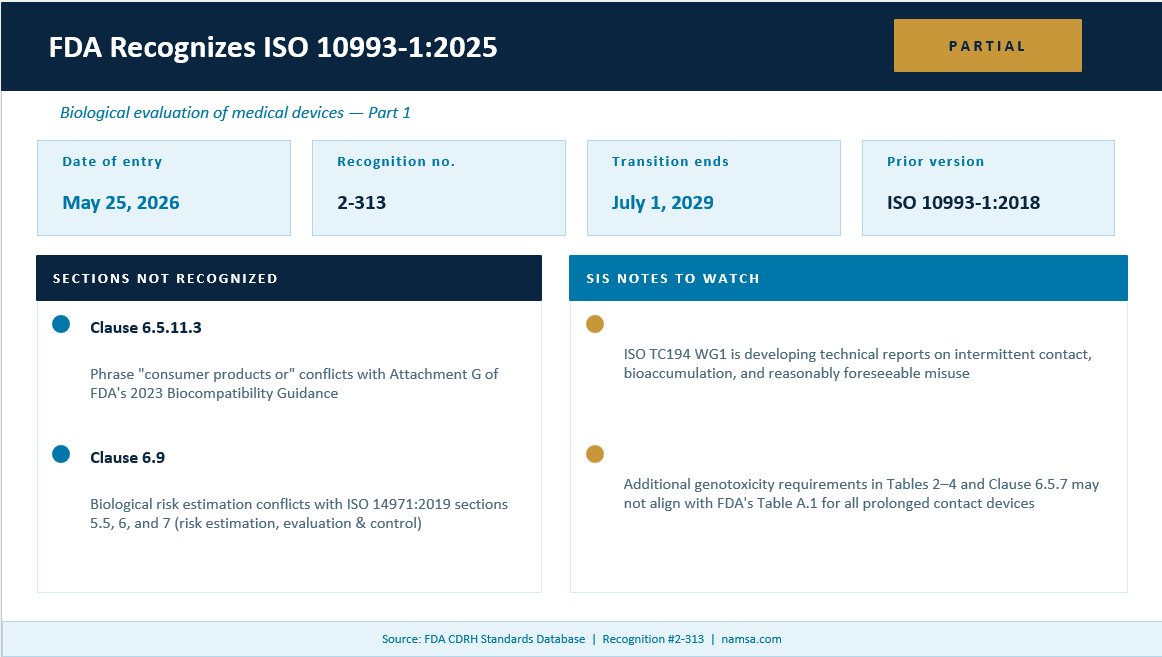
On Monday, May 25, 2026, the FDA officially recognized ISO 10993-1:2025 “Biological evaluation of medical devices – Part 1: Requirements and general principles for the evaluation of biological safety within a risk management process”. Now that it is here, what does it mean and how does it impact what biocompatibility professionals do on a daily basis?
To start with, here are some key items from the document:
Navigating the Partial Recognition
Most items are straight forward, but the extent of recognition requires you to open the Supplementary Information Sheet (SIS) and look at what is not recognized. What you will find is the following:
- Partial recognition. The following part(s) of the standard is (are) not recognized:
- Phrase “consumer products or” in clause 6.5.11.3
- Clause 6.9 Biological risk estimation
The FDA has noted that the phrase “consumer products” as used in the standard conflicts with Attachment G (Biocompatibility of Certain Devices in Contact with Skin) of the FDA’s 2023 Biocompatibility Guidance. Specifically, not all materials used in consumer products are included in Attachment G.
This phrase appears in Clause 6.5.11.3 of ISO 10993-1:2025, titled Low Risk Intact Skin Contacting Medical Devices. The relevant sentence states that biological risk may be considered low when “the skin-contacting components are made from materials with a history of safe use in consumer products or medical devices with a similar type of contact and similar or longer duration of contact.”
For submissions to the FDA, demonstrating that a material is used in a consumer product with skin contact does not automatically satisfy requirements for avoiding biocompatibility testing. Whether this approach is acceptable depends, in part, on whether the material appears in Attachment G of the FDA’s 2023 Biocompatibility Guidance. Attachment G identifies specific materials for which this approach is applicable and includes exclusion criteria.
When evaluating skin-contacting devices for FDA submission, both ISO 10993-1:2025 Clause 6.5.11.3 and Attachment G of the FDA’s 2023 Biocompatibility Guidance should be consulted.
Biological Risk Estimation and Clause 6.9
The FDA does not recognize Clause 6.9, Biological Risk Estimation, of ISO 10993-1:2025. The FDA states that this clause conflicts with Sections 5.5, 6, and 7 of ISO 14971:2019 (Medical Devices — Application of Risk Management to Medical Devices), which address Risk Estimation, Risk Evaluation, and Risk Control, respectively. The FDA has not provided additional detail in its rationale.
The practical implication is that the FDA does not recognize the biological risk estimation process as currently described in ISO 10993-1:2025. If a biological evaluation report concludes that biological risk for each applicable harm is acceptable, low, or negligible, that conclusion must be supported by evidence consistent with the FDA’s risk management framework under ISO 14971:2019.
Genotoxicity and Material-Mediated Pyrogenicity (MMP)
The FDA’s Supplementary Information Sheet (SIS) for ISO 10993-1:2025 includes two notes in the “Rationale for Recognition” section.
Note 1 informs industry that ISO TC194 WG1 is developing technical reports covering intermittent contact, bioaccumulation, reasonably foreseeable misuse, and application throughout the medical device life cycle.
Note 2 addresses a discrepancy in genotoxicity evaluation requirements. The additional genotoxicity requirements specified in Tables 2, 3, and 4, and Clause 6.5.7 of ISO 10993-1:2025 may not align with Table A.1 in Attachment A of the FDA’s 2023 Biocompatibility Guidance for all prolonged contact devices.

In ISO 10993-1:2025, genotoxicity is listed as an endpoint for evaluation for all prolonged contact device categories. In FDA’s Table A.1, genotoxicity is listed as an endpoint only for implanted devices, externally communicating devices with tissue/bone/dentin contact, and externally communicating devices with circulating blood contact — not for all prolonged contact categories.
One approach is to follow the genotoxicity evaluation requirements in ISO 10993-1:2025 Tables 2–4. This satisfies regulatory bodies outside the United States that follow the standard and avoids uncertainty about the scope of FDA’s recognition.
A related question arises regarding material-mediated pyrogenicity (MMP). Tables 1–4 of ISO 10993-1:2025 do not include MMP as an endpoint, whereas Table A.1 of the FDA’s Biocompatibility Guidance does. It is not yet clear whether the rationale provided in ISO 10993-1:2025 Clause 6.5.10.5 and Clause B.5 for omitting MMP from evaluation tables can be applied to FDA submissions. The FDA’s extent of recognition does not address this scenario in detail.
Both notes indicate that manufacturers may contact the relevant FDA review office to discuss implementation. A pre-submission meeting (Pre-Sub) would be an appropriate forum for clarifying these questions. Alternatively, a conservative approach would be to combine the applicable ISO 10993-1:2025 table with FDA’s Table A.1 and evaluate all endpoints marked with E, X, or O.
Summary
The FDA’s recognition of ISO 10993-1:2025 moves the industry closer to using the updated standard in FDA submissions. A transition period is in effect, and some manufacturers may choose to continue using the FDA’s 2023 Biocompatibility Guidance as the primary reference for biocompatibility planning during this period.