
On October 15, 2020, the U.S. Federal Food and Drug Administration (FDA) released a new draft guidance, “Select Updates for Biocompatibility of Certain Devices in Contact with Intact Skin,” which is intended to add or supersede applicable sections of the 2016 biocompatibility guidance (recently updated in 2020), “Biological Evaluation of Medical Devices-Part 1: Evaluation and Testing within a Risk Management Process,” following public comment. This document is currently being distributed for comment purposes only, and comments should be submitted within 60 days of publication.
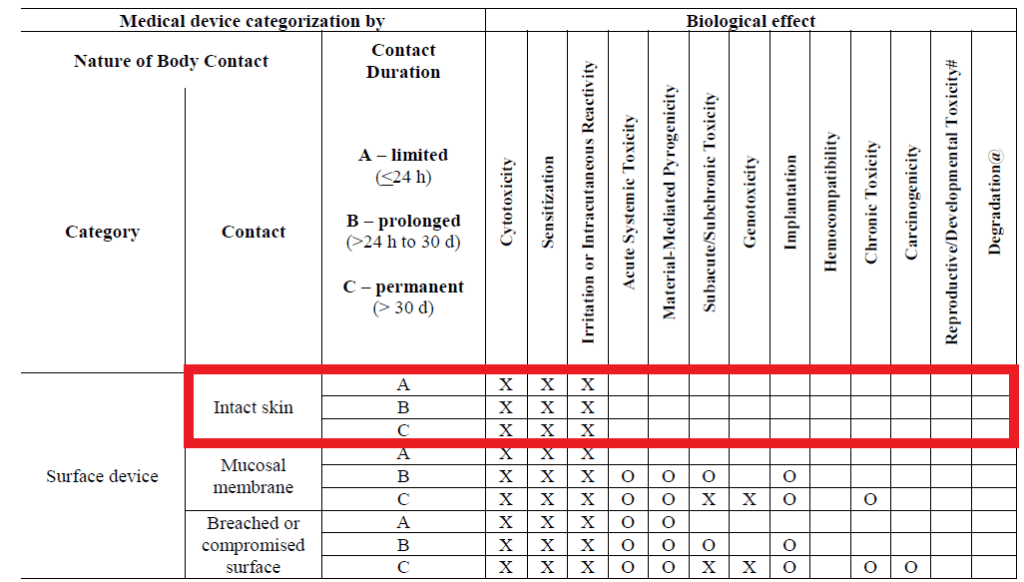
Examples of medical devices in contact with intact skin would include electrodes, external prostheses, fixation tapes, compression bandages and monitors of various types. The guidance only applies to devices in contact with intact skin and excludes other surface devices in contact with mucosal membrane and breached or compromised surfaces.
The biological endpoints for evaluation for this categorization of device include cytotoxicity, irritation, and sensitization as presented in ISO 10993-1:20181 and the FDA guidance on use of ISO 10993-1:20162 (Revised 20203) as shown below.
The FDA has determined the biocompatibility risk of various polymers and fabrics to be low based upon a safe history of use in medical devices for this categorization. The materials deemed to be low-risk include:
| Material |
| Polymers |
| Acrylonitrile butadiene styrene (ABS) |
| Cured epoxy adhesives |
| Fluoropolymers including polytetrafluoroethylene (PTFE), expanded PTFE, polyvinylidene fluoride (PVDF) and fluorinated ethylene propylene (FEP) |
| High impact polystyrene (HIPS) |
| Polyamides, including nylon |
| Polybutylene terephthalate (PBT) |
| Polycarbonate (PC) |
| Polyetheretherketone (PEEK) |
| Polyether imide (PEI) |
| Polyethylenes, including low-density polyethylene (LDPE) and high-density polyethylene (HDPE) |
| Polyethylene terephthalate (PET) |
| Polymethylmethacrylate (PMMA) |
| Polyoxymethylene (POM) |
| Polyphenolsulfone (PPSU) |
| Polypropylene (PP) |
| Polyurethane (PU) |
| Silicone |
| Fabrics |
| Polyurethane fabrics, including Lycra |
| Cotton fabrics |
| Polyamide fabrics, including nylon |
| Silk fabrics |
As these materials are considered low-risk for biocompatibility when in contact with intact skin, specific material information may be used for premarket submission rather than performing biocompatibility testing. This approach is supported by the FDA’s review experience of these materials in previous premarket submissions and aligns with the principles of the 3Rs—to reduce, refine and replace the use of in vivo testing when feasible as described in ISO 10993-2:20064. Even though the materials identified above have a demonstrated safe clinical history as defined by the FDA, there are instances where discussion with the FDA is recommended (e.g., during the Q-submission process). Situations warranting a discussion include previously failed biocompatibility results for the specific material, adverse biocompatibility-related clinical findings for the specific material, indicated use in neonates, indicated use in pregnant women or use in combination products (biological or drug). The guidance does not apply to intact skin contacting components constructed with novel materials or bulk metals, devices stored in or containing fluids or creams, in-situ cured polymers, absorbable materials or hydrogels, reprocessed single-use devices and devices containing adhesives in contact with skin.
All premarket submission for devices in contact with intact skin that meet the criteria described above should provide the following information during submission for the FDA’s consideration:
- A list of materials used to fabricate the device with direct or indirect skin contact;
- A statement (e.g., MDR analysis, literature search) confirming the listed materials have a documented history of safe use in legally US-marketed medical devices in contact with intact skin;
- A statement confirming that none of the above listed exclusions apply;
- Adverse biological responses from devices such as irritation or sensitization reactions found during the course of a clinical study, if applicable (e.g., an IDE);
- A statement indicating the manufacturer has documented in the Device Master Record how biocompatibility risks for the device have been addressed in order to prove biocompatibility testing and a detailed rationale regarding manufacturing is not necessary; and
- Precautionary labeling statements for caretakers to assess patients for adverse reactions such as irritation and sensitization to the medical device.
Following FDA clearance, it is customary for appropriate post-market surveillance to be conducted to identify any potential biocompatibility concerns for these devices. In addition, Quality Assurance (QA) controls should ensure that changes in material suppliers, materials, manufacturing processes, QA receiving specifications and QA finished device specifications do not affect the biocompatibility of the finished device. Analysis of quality data for biocompatibility concerns should also be investigated and compiled annually at minimum. Based upon the categorization of the devices, complaints related to irritation and sensitization (i.e. erythema, edema, irritation, sensitization, allergy, immune response) would be most relevant for investigation.
How Can NAMSA Help?
NAMSA is the industry leader in driving successful regulatory outcomes. Our teams of regulatory, quality, preclinical and clinical research Associates have many years of experience working for, and with, the FDA. We offer a range of biological safety solutions to ensure your medical device development program is compliant, while also focusing on the most efficient means possible to complete necessary tests.
Learn more about NAMSA’s biological safety service offerings by visiting namsa.com/services/consulting/biological-safety/. We also invite you to get in touch by submitting a request at: namsa.com/locations-contact/.
Blog References
- Biological Evaluation of Medical Devices-Part 1: Evaluation and Testing within a Risk Management Process. 2018.
- S. FDA. Use of International Standard ISO 10993-1, “Biological Evaluation of Medical Devices – Part 1: Evaluation and Testing within a Risk Management Process”. June 16, 2016.
- S. FDA. Use of International Standard ISO 10993-1, “Biological Evaluation of Medical Devices – Part 1: Evaluation and Testing within a Risk Management Process”. September 2020.
- Biological Evaluation of Medical Devices-Part 2. 2006.
