
Biological safety evaluations remain one of the most expensive and time-consuming phases of medical device development. Traditional approaches that involve full biocompatibility test panels can cost from $150k up to $500k per device and add an additional 3 to 18 months to development timelines. Costs and timeline implications are constraining device launches across the industry. Startups are forced into repeated funding cycles, while established companies face rising expenses to sustain multiple product portfolios.
The 2025 revision of ISO 10993-1 provides an opportunity to reduce this burden by reinforcing a risk-based, justification-driven approach. This standard encourages manufactures to rely on existing data, material information and toxicological assessments before completing new testing. When implemented through a robust Biological Evaluation Plan (BEP), this approach can reduce biocompatibility expenses by 40-92%, with some organizations achieving reductions as high as 92% for iterative product generations.
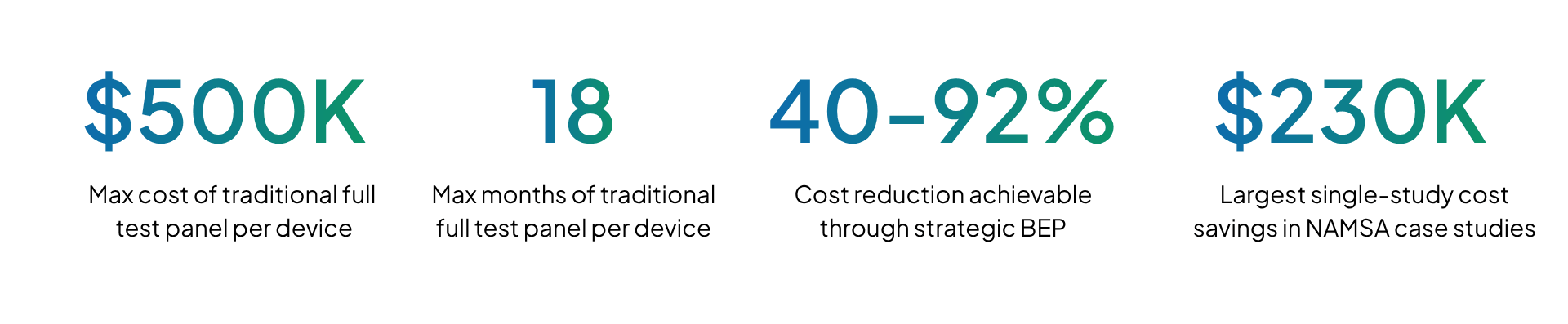
This guide outlines the regulatory, scientific, and financial mechanisms that enable these savings and provides practical guidance for constructing BEPs that minimize costs while maintaining patient safety and regulatory acceptance.
Shifting from Routine Testing to Scientific Justification
Biocompatibility testing is essential to ensure that medical devices interact safely with and within the human body. However, the historical reliance on full testing matrices, often executed by default, has created unnecessary financial and operational strains on organizations.
As device portfolios expand and development cycles accelerate, organizations are facing mounting pressure to reduce costs, shorten timelines, and eliminate redundancies, like testing. ISO 10993-1:2025 directly addresses these challenges by emphasizing scientific justifications rather than routine testing. This shift aligns with global regulatory trends and ethical priorities to reduce animal testing.
The core shift
From “test everything by default” to “test only what cannot be justified away with existing data, scientific rationale, and risk management.”
Overview of ISO 10993-1:2025: Three Principles That Enable Cost Reduction
The 2025 revision of ISO 10993-1 strengthens several key principles:
| Least Burdensome, Scientifically Valid Approach | Expanded Guidance on Leveraging Existing Data | Utilizing Robust BEPs to Mitigate Testing |
|---|---|---|
| The standard explicitly lays out that biological safety should be demonstrated using scientifically sound least burdensome methods. This shifts the default approach from ‘test everything’ to ‘test only when necessary’. | Manufacturers are encouraged to leverage all available information before beginning new studies. This information includes historical biocompatibility testing, material and chemical characterization, toxicological data, literature and predicate device information, and manufacturing/processing information. | ISO 10993-1:2025 positions the BEP as the central mechanism for documenting risk-based rationales, demonstrating data sufficiency, avoiding unnecessary testing, and ensuring regulatory alignment across submission regions. |
The 2025 revision of ISO 10993-1 also closely aligns with the 2023 FDA Biocompatibility Guidance Document Attachment G, stating that manufactures do not need to conduct full biocompatibility testing for devices made from common, historically low-risk materials that contact intact skin only. The attachment has adopted a risk-based approach, relying on historical data, type of patient contact, and material information to minimize or eliminate the need for full biocompatibility testing.
Strategic Role of BEPs
What is a BEP and Why it Matters
A BEP provides a road map for proving a device is biocompatible using a structured framework that consolidates historical data, material information, and toxicological assessments to provide testing justification and recommendations. When developed correctly, a BEP will define where biocompatibility and toxicological risks exist within a device, how to evaluate the risks, and create a test plan to mitigate risks to the patient. It can also act as a defensible document that leverages historical testing, literature reviews on device materials, and available clinical data to avoid redundant or unnecessary testing and alleviate regulatory friction.
Components of an Effective BEP
Regulatory authorities use BEPs to ensure that manufactures are not just performing tests to perform tests. Instead, regulatory reviewers want context to show that biological hazards have been identified, evaluated logically and the correct test methods have been chosen to address them. A BEP typically contains a description of the device, materials of construction, manufacturing process, type of patient contact, and contact duration. It also showcases a biological risk assessment and/or gap assessment to identify potential hazards to evaluate what information already exists and highlight what is missing. The BEP will then show a planned testing strategy in addition to scientifically sound justifications to not perform testing, where able, to ensure all biological effects required to be evaluated are considered.
Quantifying the Impact
Companies that use BEPs are not only able to reduce costs by 40-92% (depending on the type of device), but can also accelerate time-to-market and alleviate regulatory risk or additional information (AI) letters. When developed properly, a robust BEP can speed up development and, in turn, save manufactures weeks and/or months of testing timelines, getting the device to market quicker.
How NAMSA Can Help
NAMSA prepares BEPs and Biological Evaluation Reports (BERs) that are accepted by FDA reviewers and EU Notified Bodies. Our consultants have authored BEPs across all device classes, contact types, and risk profiles, with a track record of successfully justifying testing waivers that save manufacturers significant time and cost.
Case Studies: Demonstrated Cost Reduction and Regulatory Success
Case Study 1: Optimizing Large Animal Study to Reduce Testing Cost for an Investigational Device Exemption (IDE) application for an Early Feasibility Study (EFS)
| DEVICE & CHALLENGE | BEP APPROACH & METHODOLOGY |
|---|---|
| Complex cardiovascular device with delivery systems and multiple components, varying in patient contact from limited (≤24 hr.) to prolonged (>7 days), with direct and indirect contact with circulating blood and intact/breached surface. Standard test plan: Chemical Characterization, Toxicological Risk Assessment, Cytotoxicity, Sensitization, Irritation, Acute Systemic Toxicity, Pyrogenicity, Subacute/Subchronic Systemic Toxicity, Genotoxicity, Implantation, Hemolysis, Coagulation, Complement Activation, Thrombogenicity. | Leverage historical data on materials of construction, optimize the large animal study to include evaluation of required biological effects and strategically plan the EFS to cover additional endpoints. – Paper-based Material Characterization in lieu of Chemical Characterization and TRA – Large Animal Study used to address Subacute/Subchronic Systemic Toxicity, Thrombogenicity, and Hemolysis – EFS indicators to address Pyrogenicity and Complement Activation Testing recommendations as outlined through a risk-based approach BEP: Large Animal Study, Cytotoxicity, Sensitization, Irritation, Acute Systemic Toxicity only |
| ~$230,000 saved 58.7% cost reduction, testing scope cut from 14 required endpoints to 5 through strategic data leverage and study consolidation | |
Case Study 2: Iterative Product Generation(s) and Material Changes
| DEVICE & CHALLENGE | BEP APPROACH & METHODOLOGY |
|---|---|
| Class II Gastrointestinal Tube and Accessory with limited (≤24 hr.) contact with mucosal membrane via indirect drug contact. Standard test plan: Chemical Characterization, Toxicological Risk Assessment, Cytotoxicity, Sensitization, Irritation. | Perform targeted gap analysis on material change. Leverage predicate device data and material manufacturer testing on the new material. – Historical Chemical Characterization data and Material Characterization on the changes – Limited (≤24 hr.) exposure duration to mitigate toxicological risk – Biocompatibility re-testing omitted based on well-established material history, material manufacturer biological/physical data, predicate device testing, and limited exposure duration Testing recommendations as outlined through a risk-based approach BEP: No additional testing required |
| ~$120,000 saved 92% cost reduction, all planned testing eliminated through strategic use of historical data, predicate leverage, and limited exposure justification | |
Case Study 3: Intact Skin Contacting Device under FDA’s 2023 Biocompatibility Guidance Document Attachment G
| DEVICE & CHALLENGE | BEP APPROACH & METHODOLOGY |
|---|---|
| Device accessories categorized as limited (≤24 hr.) but repeated intact skin contact — classified as long-term (>30 days) under ISO 10993-1. Standard test plan: Cytotoxicity, Sensitization, Irritation. | Use of established materials and FDA Attachment G justifications. – Intact skin contact device comprised of low-risk, historically safe materials – No additives or processing steps that introduce risk or alter material safety – Post-Market Surveillance data demonstrating safety history with minimal biocompatibility incidents – Realistic cumulative exposure assessment demonstrates low patient risk Testing recommendations as outlined through a risk-based approach BEP: No additional testing required |
| Testing fully eliminated Cost reduction for manufacturing, avoidance of lab dependency delays, timeline acceleration, faster submission approval | |
Financial and Operational Benefits Beyond Cost Reduction
A well-written BEP can offer advantages that extend well beyond eliminating unnecessary testing and reducing cost. By proactively establishing a clear evaluation strategy, organizations can streamline development activities, reduce rework and shorten time-to-submission.
- Accelerated Market Entry: This structured approach allows developers and manufacturers to focus only on necessary testing, which accelerates market entry and minimizes delays caused by late-stage regulatory questions and requests.
- 3R Alignment: BEPs also support global regulatory efforts to reduce animal testing by prioritizing non-animal methodology and avoiding redundant in-vivo studies, which aligns organizations with the evolving ethical and regulatory projections.
- Global Regulatory Alignment: A robust BEP reduces the likelihood of AI requests by helping reviewers understand how each endpoint was evaluated — aligning ISO 10993-1:2025 justifications with FDA, EU, and global submission requirements.
- Reduced Lab Dependency: BEPs decrease reliance on laboratories by shifting an emphasis toward literature and historical data. This shift creates an important advantage in budgetary and timeline predictions, especially given testing backlogs and laboratory capacities.
- Better R&D Capital Allocation: By eliminating low-value testing and enabling earlier, more informed decisions about device biocompatibility, BEPs can improve R&D capital allocation. These efficiencies could scale across manufacturing portfolios, allowing organizations to direct resources towards programs with the strongest commercial potential.
Practical Guidance for Implementing BEP-Driven Strategies
- Start with a defensible device narrative. A successful BEP begins with a defensible narrative that requires a detailed understanding of the device materials, manufacturing process, and exposure scenarios based on its intended use. Showcasing clear consideration between device characteristics and biological effects can strengthen the rationale for when testing is able to be justified or leveraged.
- Involve toxicologists early. Early involvement of toxicologists can ensure the plan reflects both the scientific rigor and regional regulatory expectations for a successful submission.
- Structure data to support conclusions. A robust and thorough BEP should be structured in a format that allows the data to support the conclusion. Consistent terminology and documentation throughout a BEP should be used to show supporting evidence in a clear and concise manner.
- Avoid common pitfalls. Incomplete device categorization or reliance on outdated data can be avoided through early planning and cross-functional coordination.
- Plan for portfolio-wide application. BEP strategies should be designed with iterative product generations in mind. The savings in Case Study 2 (92%) were achievable precisely because the original BEP was built in a way that made material change justifications straightforward, rather than requiring a full re-evaluation each time.
Conclusion
ISO 10993-1:2025 provides a clear pathway for organizations to streamline their biological evaluation programs through well-developed BEPs. By consolidating historical data, material information, and toxicological insights into a single, justification-driven framework, BEPs allow manufacturers to demonstrate biological safety while avoiding unnecessary testing and increased costs and delays that come with it.
These results are not only tangible but can be significant. As shown in the case studies, BEP-driven strategies can reduce testing costs anywhere from 40-92%, shorten development timelines, and minimize regulatory pushbacks. These gains translate directly into faster market access, more predictable submissions and better use of R&D capital. BEPs also reduce dependency on laboratories, which is becoming increasingly important as backlogs and timelines continue to grow.
For organizations managing expanding device portfolios and tighter development cycles, BEPs offer a scalable, high ROI approach that strengthens both scientific rigor and business performance. Companies that utilize BEPs early in development will be better positioned to control costs, accelerate innovation and maintain a competitive edge in a rapidly evolving regulatory environment.
Frequently Asked Questions (FAQs)
Can a BEP really eliminate all biocompatibility testing, or is there always some testing required?
Yes, in the right circumstances, a BEP can eliminate all new biocompatibility testing. However, this outcome depends on several conditions being met:
- Well-characterized, historically safe materials with strong existing data
- Availability of predicate device data or material manufacturer safety data
- Post-Market Surveillance data demonstrates an established safety history
For complex, high-risk, or novel-material devices, some targeted testing is typically still required, but the BEP ensures only the minimum necessary tests are conducted, and that each test is selected for a specific scientific reason rather than by default.
How does ISO 10993-1:2025 differ from the 2018 version in terms of BEP requirements?
The 2025 revision aligns and integrates with ISO 14971 risk management principles into the biological evaluation framework. Key differences that affect BEP strategy include:
- Stronger emphasis on justification over testing: The 2025 version is more explicit that manufacturers must actively justify why testing is or is not needed, not simply plan to test against all Annex A endpoints as a default.
- Expanded guidance on existing data leverage: More detailed direction on how to use chemical characterization, toxicological risk assessments, clinical history, and literature as evidence in place of new testing.
- Alignment with FDA Attachment G: The 2025 standard directly aligns with the intact skin device exemptions introduced in the 2023 FDA Biocompatibility Guidance
Will a BEP that reduces testing be accepted by both the FDA and EU Notified Bodies?
Yes, when prepared correctly. Both the FDA and EU Notified Bodies review BEPs as primary regulatory documents. The key is that a BEP must provide:
- A clear, logical risk-based assessment that demonstrates hazards have been identified and evaluated systematically
- Transparent documentation of all data sources used to support waived tests with robust justifications
- Consistent terminology and cross-referencing that allows a reviewer to follow the rationale without ambiguity
Regulatory reviewers use BEPs to confirm that manufacturers are not simply avoiding testing without justification. A well-constructed BEP with documented scientific rationale is more likely to reduce AI requests than a standard test matrix because it demonstrates that every endpoint was actively considered.
How does FDA Attachment G work for intact skin devices and what are its limits?
Attachment G of the FDA’s 2023 Biocompatibility Guidance allows manufacturers to waive full biocompatibility testing for devices that contact intact skin only, provided they meet all the following criteria:
- Device components are made from materials with a history of safe use in medical devices or consumer products with similar skin contact (note: ISO 10993-1:2025 partially aligns here but with a nuance, the FDA does not fully recognize the “consumer products” reference for its submissions)
- No known adverse biocompatibility history for the materials
The limits of Attachment G are important: it applies only to intact skin contact. Devices with mucosal, breached skin contact, or implantable, or those using novel materials, are outside its scope and require a conventional BEP with appropriate evidence. The FDA’s May 2026 recognition of ISO 10993-1:2025 also introduced a specific partial exclusion related to the “consumer products” language in Clause 6.5.11.3. Manufacturers should consult the Supplementary Information Sheet alongside the standard for FDA submissions.
