
Introduction
The European Union’s (EU) In Vitro Diagnostic Regulation (IVDR) of Medical Devices 2017/746 is drastically impacting the IVD industry at varying levels. Under this directive, all products—irrespective of class—that stay on the market following the transition date of 26 May 2022* must be reassessed for IVDR compliance to ensure that products are fit for purpose and safe for use. There is no automatic pass granted based on the time that a product has been on the market, and therefore, no ‘grandfathering’ is permitted. Notified Bodies and manufacturers will now have to prepare for this as the industry moves to an environment of greater control and oversight. (* The date of compliance for certificates issued under the In Vitro Diagnostic Directive [IVDD] is 27 May 2024.)
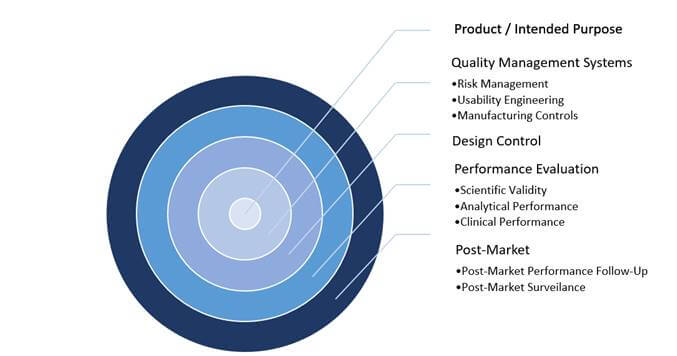
Figure 1: Critical Elements within IVD Product Development Pathway
The fundamental premise of the IVDR and IVDD are the same, as both outline that manufacturers must:
- Develop products based on market need;
- Develop an intended purpose; and
- Control the design, development and performance evaluation of the device while managing risks and associated mitigation to ensure safety, effectiveness and suitability for intended purposes.
The above information must be evidenced in a Technical File which may need to be submitted to a Notified Body for review (dependent on device classification) to enable CE marking and market access. However, the IVDR has introduced changes that will affect how products are developed and managed on the market; these critical elements are highlighted in Figure 1 above.
Planning for IVDR Compliance
IVD manufacturers are familiar with structured systems in the development and management of products. However, it is becoming clear that in order to develop and manage a product in accordance with the IVDR, there must be greater emphasis on, and appreciation of, the supporting processes that provide compliance evidence. Consequently, regulatory strategy, management support and QMS effectiveness have become pillars of conformity (e.g. a strong and effective QMS will drive the required control around Unique Device Identification [UDI] labelling and/or the reporting of post-market safety information).
Product / Intended Purpose and Performance Evaluation
It is well understood that a product’s intended purpose is central to the development of a device. However, the IVDR is far more explicit in its requirements regarding Sponsors’ intended purpose statements. In addition to a product’s use (specimen type, quantitative or qualitative data, automated or semi-automated information, testing population and intended users), the intended purpose must demonstrate details of a product’s function and what is to be detected. In a typical scenario, this includes the detection of the biomarker/analyte, the specific disorder, condition or risk factors that are to be identified; this not only helps ensure a solid foundation for development, but it also provides important data to the end user.
The intended purpose flows through the product development lifecycle and is then brought into a new process of “performance evaluation,” which is not just simply analytical verification. This process combines the scientific validity of the analyte with the clinical condition, analytical performance and an assay’s ability to provide a clear result to the clinical condition/ pathological process/intended state. Following, these are laid out in a Performance Evaluation Plan (PEP), and ultimately in a Performance Evaluation Report (PER).
Maintaining Market Availability
Getting a product to the global marketplace is often a challenge, and securing a CE mark to sell a product in Europe is generally good news for all involved. Something less glamorous, but arguably more critical, is keeping that product on the market and ensuring that it continues to be “state of the art” and safe throughout its lifetime. Post-market activities, as outlined within the IVDR, are a major step up from the requirements of the outdated IVDD. There are new processes, including Post-Market Performance Follow-Up (PMPF), Periodic Safety Update Reporting (PSUR) and Post-Market Surveillance Planning and Reporting (PMSP/PMSR), all with an emphasis on the product lifecycle.
The days of marketing a product and simply tracking complaints are gone. The continuous and systematic review of relevant data on product quality, performance and safety throughout a device’s lifetime ensures that the product continues to perform as intended. This continued review, through data generated via the company QMS, must feed in to the Technical File. Notably, the performance evaluation documentation must be updated according to product classification and the PMSR with any new clinical evidence and published literature supporting a product’s continued use.
IVDR Compliance Roadmap
NAMSA, in collaboration with hundreds of global IVD manufacturers, has developed a proven roadmap that identifies key areas for IVDR compliance and allows clients to enter this changing regulatory environment with confidence. The areas outlined below in Figure 2 require significant effort and investment, and are critical in a manufacturer’s ability to continue to sell IVD products in the EU. Following a roadmap, or a well thought-out, organized path forward, will enable easier compliance and market access.
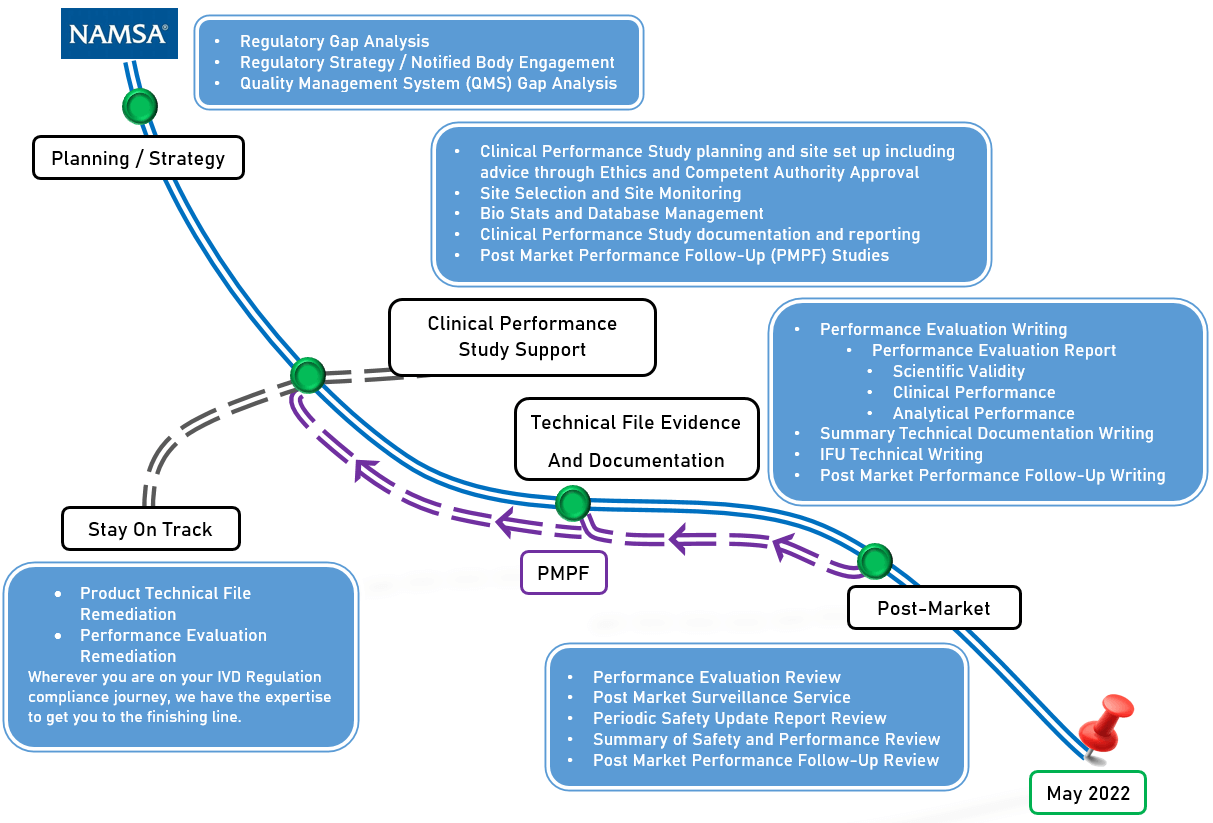 Figure 2: IVDR Compliance Roadmap
Figure 2: IVDR Compliance Roadmap
Helping IVD Sponsors Bridge the Gap
Outlined below are two abbreviated case studies for purposes of this blog, which highlight NAMSA’s unique roadmap process.
CASE STUDY I
Client Objective:
NAMSA partnered with a large multinational company in the field of molecular diagnostics. The task was to provide a Performance Evaluation for a Real Time Polymerase Chain Reaction (qPCR) kit.
Challenge:
The performance evaluation determined a gap related to the State of the Art (SOTA) in which the product did not have a complete detection profile in line with current industry guidelines (and in comparison with similar devices on the market). NAMSA recommended that this gap be addressed through PMPF in order to bring the product in compliance with SOTA.
Strategic Approach:
In collaboration with this client, NAMSA:
- Performed an assessment of the product’s intended purpose following IVDR requirements
- Carried out a performance evaluation with a PEP that highlighted the intended purpose, SOTA and product risk profile, as well as the evaluation of multiple source data (for the PER)
- Performed objective and systematic literature searches to establish the scientific validity of the analyte and the SOTA in medicine
- Performed a systematic evaluation of the data to support the analytical and clinical performance of the product and to determine the benefit-risk ratio
- Compared the collected clinical evidence against SOTA to identify potential gaps in the performance of the product
- Provided recommendations that gaps be addressed via PMPF activities
Outcome:
Benefits realized through NAMSA’s work:
- The client performed this work without the need to recruit additional internal resources in-house, thereby reducing costs
- A significant gap was quickly identified that was a risk to end users and was controlled
- Recommendations were brought forward to bring the client’s product back into conformity prior to submission, potentially reducing Notified Body review time
CASE STUDY II
Client Objective:
NAMSA collaborated with a large Japanese manufacturer looking to broaden their market reach by entering the European Market. The task was to review existing clinical evidence and provide an analysis of the gaps and to conduct a performance evaluation for submission to a Notified Body.
Challenge
Central to this work was managing the regulatory and cultural differences between Europe and Japan. The clinical evidence was in Japanese and there were gaps in data that would be required for European submission; most notably European clinical data were missing.
Strategic Approach
In doing this work NAMSA:
- Formed a project structure to ensure the best possible communication between the Japanese client and EU entities, and to establish trust
- Reviewed the clinical evidence (PER) and performed a gap analysis of the provided data, following IVDR requirements
- Provided a remediation plan for the required analytical data and gaps in clinical performance data, specifically related to the target EU population
Outcome
Benefits realized through NAMSA’s work:
- The client managed to perform this work without the need to recruit further internal resources. thus reducing overall costs
- NAMSA provided quick, accurate information and identified gaps in analytical and clinical performance data
- NAMSA swiftly created a plan, in partnership with the client, to ensure proper remediation and market access strategies and approaches
- The client’s written PERs were accepted
- Route to EU market was accelerated, which led to quicker revenue realization
Conclusion
Since the IVDR was published in 2017, there has been much information published to assist Sponsors in tackling the changes set forth in the new regulation. However, there have been significant challenges along the compliance pathway: from the designation of Notified Bodies to regulatory authorities providing little guidance on expectations; but, manufacturers must still find a way to plan for these changes and identify gaps to prioritize these areas to meet compliance.
How Can NAMSA Help?
NAMSA’s dedicated team of IVD specialists and scientists not only understand how to guide IVD manufacturers through clinical and regulatory requirements, but we also help Sponsors simplify the development and implementation of effective development strategies. Whether supporting IVD regulatory assessments and submissions, developing IVDR compliant technical files, designing and managing clinical trials or building ISO 13485:2016 and 21 CFR part 820 compliant quality systems… we’ve got you covered. Learn more about NAMSA’s IVD solutions by clicking here.
