
Medical Dictionary for Regulatory Activities (MedDRA) is a standardized, clinically validated international medical terminology developed under the backing of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). It is widely used in clinical trials, particularly for medical devices and pharmaceuticals, to ensure consistent and accurate reporting of adverse events and other medical information.
What is MedDRA Coding?
MedDRA coding is used by Sponsors and regulatory authorities in the medical device and pharmaceutical industry. It facilitates the exchange of clinical information through standardization, supporting coding (data entry), retrieval, and analysis of clinical information about human medical products, including pharmaceuticals, medical devices, biologics, vaccines, and drug-device combination products.
History and Purpose
MedDRA was first implemented in 1999, primarily in Europe, Japan, and the US. Its creation was driven by the need for a standardized terminology that could be used globally to facilitate the sharing of regulatory information for medical products. The terminology is used throughout the entire regulatory process, from pre-marketing to post-marketing, and for data entry, retrieval, evaluation, and presentation.
How is MedDRA Coding Used?
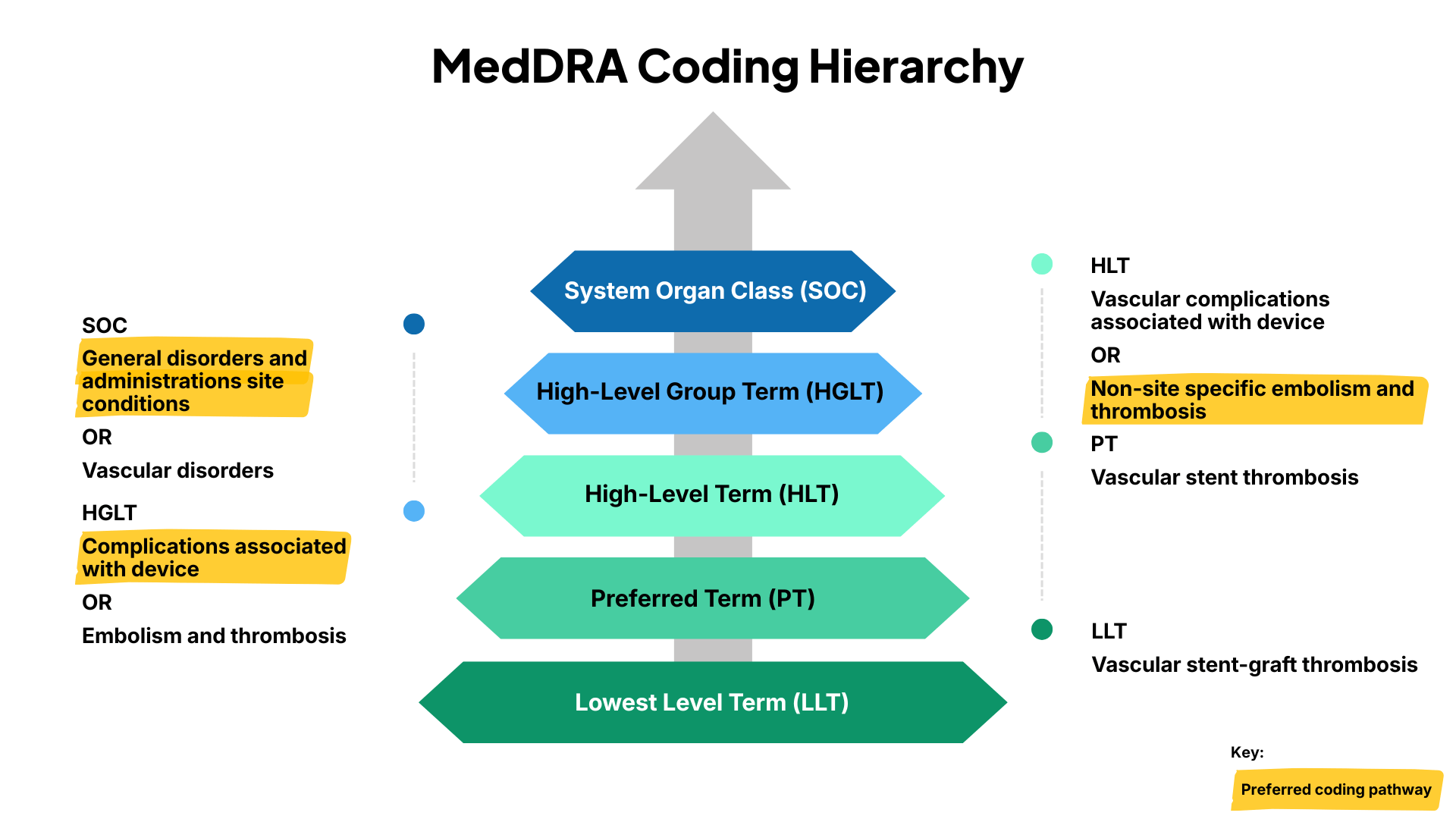
MedDRA supports the coding of adverse events (AE), medical history, and other relevant medical information. The terminology is structured into five levels:
- System Organ Class (SOC)
- High Level Group Term (HLGT)
- High Level Term (HLT)
- Preferred Term (PT)
- Lowest Level Term (LLT)
Each adverse event is coded into a LLT, which in the dictionary is linked to one PT. Multiple LLTs can code into one PT. An example is the PT “Myocardial infarction” which contains the LLTs “Heart attack”, “Myocardial infarct”, “Myocardial infarction”, “Inferior MI”, and many more. The PT then corresponds to one or more HLT. The HLT is linked to one specific HLGT and that is linked to one SOC. This multi-axiality allows a single AE term to be coded into multiple SOCs.
MedDRA provides a primary SOC for each PT, which can be important depending on the etiology of the adverse event or the therapeutic area of the medical device. An example would be the LLT “Vascular stent-graft thrombosis” which can be coded into both the “Vascular disorders” and “General disorders and administration site conditions”. Both can be used, but it is important that similar adverse events are coded consistently into the same SOC, ensuring efficient analysis and comparability. The “General disorders and administration site conditions” uses the HLGT “Complications associated with device” indicating a relationship between the adverse event and the device.
This hierarchical structure allows for detailed and specific coding of medical information. As mentioned above, to ensure consistency in coding, Sponsors should develop specific guidelines for each clinical trial. These guidelines include instructions on how to handle misspellings, abbreviations, acronyms, combination terms, and ambiguous terms. Additionally, it is important that AE Case Report Forms (CRFs) are queried consistently, to ensure that proper AE terms are being used throughout the study.
MEdDRA Coding of Device-Related Issues
MedDRA coding can apply to medical device-related issues. While MedDRA is not a device dictionary (i.e., it doesn’t code for specific device names or models), it does support the coding of medical conditions, adverse events, and product issues that may be associated with medical devices.
What MedDRA Can Code for Medical Devices:
- Device-related adverse events (e.g., “device malfunction,” “implant site infection”)
- Procedural complications (e.g., “Post procedural pain”)
- Product quality issues (e.g., “device breakage,” “battery failure”)
- Use errors (e.g., “incorrect device usage”)
- Injury or poisoning due to device (e.g., “burn from defibrillator”)
Benefits and Limits of MedDRA Coding
Benefits of MedDRA Coding
The primary benefit of MedDRA coding is its ability to standardize medical terminology, which facilitates the exchange of clinical information between different trials, and improves the accuracy of data reporting. This standardization is crucial for product evaluation and supports the identification and assessment of safety signals, which is essential for materiovigilance.
Limits of MedDRA Coding
Despite its benefits, MedDRA coding has limitations. One significant challenge is the complexity of the terminology, which can lead to inconsistencies in coding if not used correctly. The hierarchical structure, while detailed, can sometimes result in ambiguity, especially when coding complex medical conditions or events. Additionally, MedDRA requires regular updates to keep pace with evolving medical knowledge and practices. It is recommended that the dictionary version chosen at the start of a trial is used throughout the trial. Major changes in public health, such as the COVID-19 pandemic, can be seen as a reason to change the dictionary to ensure that these events are adequately coded.
Regulatory Authorities and Guidelines
Regulatory authorities such as the FDA, European National Competent Authorities (NCA), and Asian authorities recognize and endorse the use of MedDRA coding. To ensure MedDRA has interoperability with other terminologies that are used in clinical trials and in regulatory reporting, the Maintenance and Support Services Organization (MSSO) has developed necessary mappings between MedDRA and said terminologies. These include, but are not limited to, Common Terminology Criteria for Adverse Events (CTCAE) grading, International Medical Device Regulators Forum (IMDRF) terminology, and World Health Organization’s Adverse Reaction Terminology (WHO-ART).
Guidelines such as ISO 14155, which outlines good clinical practice for medical device trials, recommend the use of standardized terminologies like MedDRA for adverse event reporting. The ICH M1 Points to Consider Working Group develops and maintains documents on the use of MedDRA for data entry (coding) and data retrieval/analysis, ensuring its consistent application across different regions.
IMDRF Coding Dictionary
The IMDRF has developed its own coding dictionary for adverse event reporting, known as the IMDRF Terminologies for Categorized Adverse Event Reporting. This dictionary includes terms for medical device problems, investigation types, findings, conclusions, health effects, and medical device components. The IMDRF coding dictionary aims to harmonize adverse event reporting across different regulatory jurisdictions, improving the consistency and reliability of data.
IMDRF vs. MedDRA: Pros and Cons
Pros of IMDRF:
- Specificity for Medical Devices: IMDRF coding is tailored specifically for medical devices, addressing unique issues and components that may not be covered comprehensively by MedDRA.
- Global Harmonization: IMDRF aims to harmonize adverse event reporting across different regulatory jurisdictions, which can facilitate international regulatory processes.
- Flexibility: The IMDRF dictionary is designed to be adaptable to various types of medical devices and their specific needs.
Cons of IMDRF:
- Limited Scope: While IMDRF is excellent for medical devices, it may not be as comprehensive for pharmaceuticals and biologics compared to MedDRA. The number of available terms is notably lower than in the MeDRA dictionary.
- Adoption and Training: Transitioning from MedDRA to IMDRF requires significant training and adaptation, which can be resource-intensive.
Pros of MedDRA:
- Comprehensive Coverage: MedDRA covers a wide range of medical products, including pharmaceuticals, biologics, and combination products.
- Established Use: MedDRA is widely adopted and recognized by major regulatory authorities globally, ensuring consistency in reporting.
Cons of MedDRA:
- Complexity: The detailed hierarchical structure can lead to inconsistencies if not used correctly.
Conclusion
MedDRA coding plays a crucial role in MedTech and medical device clinical trials by standardizing medical terminology and facilitating the exchange of clinical information between different trials and devices. Its widespread adoption by regulatory authorities and endorsement by guidelines such as ISO 14155 highlight its importance in ensuring accurate and consistent reporting of adverse events. While alternatives like the IMDRF coding dictionary are available, MedDRA remains the preferred choice for many regulatory processes due to its specificity and comprehensive coverage.
Frequently Asked Questions (FAQs)
Is MedDRA Coding mandated for my clinical trial?
No, while MedDRA is not legally required by the FDA or other national regulatory authorities, it is preferred. During quarterly or annual safety reporting to the authorities, MedDRA coding of adverse events provides a more systematic and comprehensive approach, making it easier for both the Sponsor and authority to identify safety signals.
Do I need to be MedDRA certified prior to coding adverse events?
No, a certification is not a requirement for coding adverse events but is highly recommended. A MedDRA coding certification provides an in-depth examination on complex coding, including coding of adverse event narratives. This allows one to better understand when queries are required to ensure adequate reporting of adverse events. This ensures consistent coding approaches from PT to SOC throughout different device and drug trials and programs. At NAMSA, all safety associates who perform MedDRA coding are certified coders.
If my CRO already has a MedDRA subscription, am I, as a Sponsor, still required to obtain one?
A MedDRA subscription is required to access the tools needed for coding and to use the coding for regulatory purposes. Even when a CRO is providing this service, a Sponsor is required to possess a MedDRA subscription. The prices for subscriptions vary and are based upon annual revenue. Subscriptions for regulatory authorities and non-commercial or non-profit organizations are free.
