
Table of Contents
- Why Human Factors and Usability Testing is Important
- Incorporation of HF/UE in Medical Device Regulations
- User Interface Design/Evaluation
- Conducting Adequate Formative Evaluations
- Simulated Use Testing (Validation)
- Design and Interpretation of Summative Test Results in Simulated Use Validation Studies
- Human Factors and Usability Testing as a Competitive Advantage
- Conclusion
- Frequently Asked Questions (FAQs)
- Resources and References
Usability engineering, often used interchangeably with human factors, focuses the design of user interfaces (UIs) that support efficient and intuitive interaction, rapid learning, and user satisfaction. Usability is a multi-dimensional concept describing how easily and reliably users can interact with a system or product to achieve intended outcomes. Human Factors (HF) and usability engineering for medical devices aim to minimize use-related risks and errors to ensure the device is safe and effective. Usability testing is no longer optional in medical device development. It is a critical contributor of patient safety, regulatory compliance, and commercial success. Regulatory authorities around the world increasingly expect manufacturers to demonstrate that their devices can be used safely, effectively, and intuitively by intended users in real- world environments.
Despite these expectations, many medical device manufacturers, particularly startups and emerging companies, experience challenges integrating human factors activities into the development lifecycle in a way that is both efficient and sustainable.
Understanding the purpose, timing, and regulatory value of human factors testing is essential to establishing a compliant, scalable, and cost-effective strategy.
Why Human Factors and Usability Testing is Important
Adverse events over the past several decades have shown disturbing trends in post-market events that are attributable to design issues regarding the user interface (UI) of medical devices. Infusion pumps, automatic electronic defibrillators, ventilators, and combination products such as drug auto-injectors, have a history of use-related design problems resulting in overdoses, improper delivery, incorrect diagnosis and dangerous delays in therapy. As part of the systematic process to reduce errors by regulatory bodies, medical device companies in the US and EU have been introduced to the disciplines of Human Factors and Usability Engineering (HF/UE). HF/UE has been applied in the automotive, aerospace, and telecommunication industries for more than 80 years, but became formally integrated into the medical device industry during the 2000s, with a major regulatory push by the FDA in 2011 and final robust enforcement beginning around 2016.
Human factors and usability testing is crucial because it ensures products – particularly medical devices and software – are safe, effective and intuitive by evaluating how people actually interact with them. Human Factors engineering focuses on the interaction between users, devices, and environments. Usability has a major impact on healthcare, particularly with regard to the overall effectiveness of medical devices. Simply put, if usability is lacking, the completion of user tasks may be slower and more error prone. Therefore, the delivery of therapy will suffer, and patient safety may be compromised. It’s well known that easy-to-use products are more popular, resulting in market discrimination and competitive advantage. Therefore, usability can be a positive attribute from a business and sales perspective in addition to controlling risk.
Incorporation of HF/UE in Medical Device Regulations
In response to the increasing number of use-related adverse events associated with UI design, the US Food and Drug Administration (FDA) has integrated HF/UE reviews as a routine component of the premarket approval process. Within the Center for Devices and Radiological Health’s (CDRH), these reviews are conducted by the Office of Product Evaluation and Quality (OPEQ) and are guided by the FDA’s published guidance, Applying Human Factors and Usability Engineering to Medical Devices.
Similarly, international regulatory authorities have adopted IEC 62366-1, Medical devices – Application of usability engineering to medical devices, as a part of their regulatory frameworks outside of the United States. Both the FDA guidance and IEC 62366-1 define a structured usability engineering process that spans the device development lifecycle and culminates in usability validation testing of the final user interface design under simulated use conditions.
Basic Activities in the HF/UE Process and Alignment with Device Risk Assessment
Human Factors and Usability Engineering processes involve defining user groups, conducting task analysis, performing formative evaluations, and executing summative usability validation testing to ensure safe, effective device use. These activities align directly with medical device risk management process defined by ISO 14971 by identifying, mitigating, and validating controls for critical user-related hazards such as those documented in a Use-Related Risk Analysis (URRA).
Biggest Challenges for Device Manufacturers
The following challenges remain in meeting the intent of the FDA guidance and IEC 62366-1:
- Conducting adequate formative evaluations prior to final design validations
- Conducting and documenting comprehensive use-risk assessments
- The design and interpretation of summative test results into simulated use validation studies
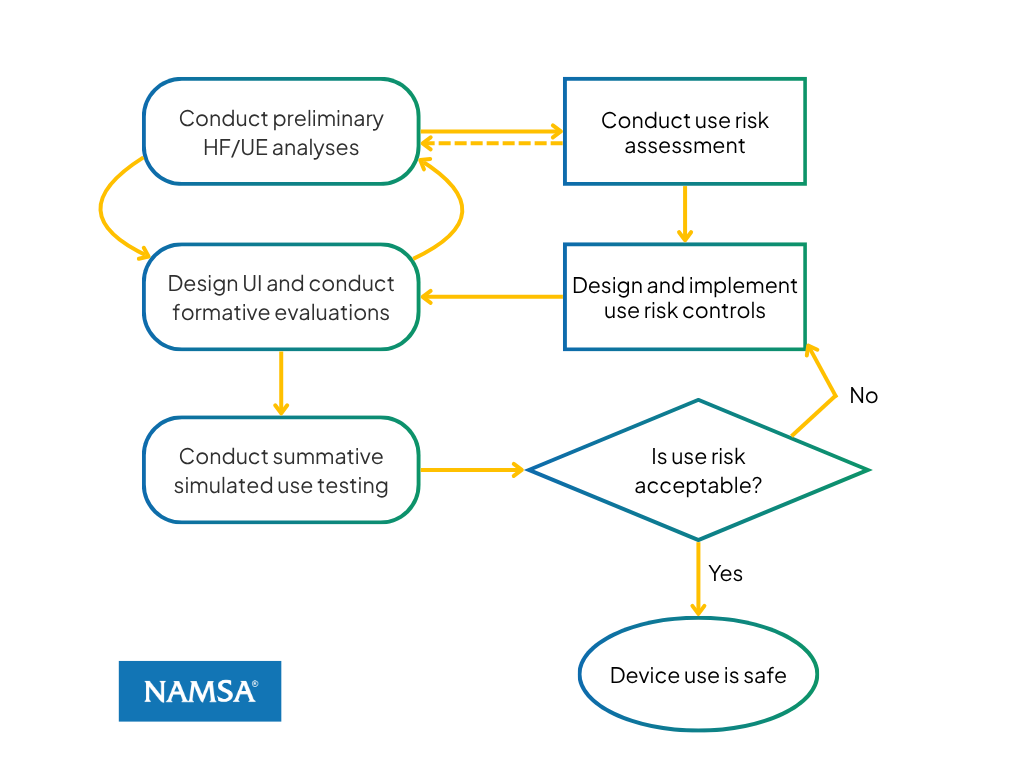
Figure 1: Relationship of HF/UE process flow with ISO 14971 risk control process
Figure 1 illustrates the relationship between the major phases of HF/UE and the corresponding stages of device risk assessment and management. On the left are the phases of HF/UE activities, while the right side shows the stages of risk assessment and management showing how these processes relate to each other. The relationship between HF/UE process and ISO 14971 can be summarized as:
- Risk Assessment: ISO 14971 risk analysis is supported by early HF/UE activities characterized by performing a preliminary analysis focused on understanding users, their use environment, interaction tasks, and potential use-related hazards associated with the device interface.
- Risk Control Implementation: The implementation of risk controls corresponds to user interface design and evaluation, including the testing of interface features intended to mitigate identified use-related risks.
- Residual Risk Evaluation: Acceptance of risk controls is addressed through simulated use testing, which evaluates whether residual patterns of use error on critical tasks remain and whether use-related risks have been reduced to an acceptable low level.
This alignment ensures that HF/UE activities are fully integrated into the overall device risk management framework and provide objective evidence supporting use safety.
User Interface Design/Evaluation
Both the FDA and IEC 62366-1 emphasize a user interface UI design process driven by iterative formative evaluations. These evaluations are conducted early and throughout the design development to improve usability and manage use-related risks. Formative evaluations serve two primary objectives: enhancing the intuitiveness and ease of use of the device, and identifying and mitigating potential use-related hazards. In some cases, these objectives may create trade-offs, as increased simplicity does not always align with safety requirements.
Several established resources support formative evaluation during early device UI development. AAMI HE75, Human factors engineering – Design of Medical Devices, (Clause 9), provides practical guidance on usability testing methods such as cognitive walkthroughs, heuristic evaluations, and walk-through-talk-through studies. IEC 62366-1, Annex D, also describes these techniques in the context of usability engineering.
Interface design standards and best practice references such as HE75 provide excellent guidance for the design of displays, controls, software graphical UIs, alarms, surgical tools, instructions for use (IFU), and other elements of the UIs. However, HE75 stresses the value of interactive user testing during the course of UI design. The role of formative techniques such as cognitive walkthroughs, heuristic evaluations, and walk-through-talk-through usability tests, are essential for obtaining actionable user feedback to the design team regarding both ease of use and effective use error mitigation.
Conducting Adequate Formative Evaluations
Although formative evaluations typically require modest effort and resources, many manufacturers limit or bypass these activities during user interface development. This is often driven by common misconceptions, including the following:
- Perceived Cost and Time Burden: Formative studies are generally efficient and can be completed with a small number of representative users. When conducted early in the design process, studies involving as few as 8-10 participants can identify 90% of existing design flaws enabling timely design improvements that reduce errors and use-related problems. As the designs matures, formative evaluations can also serve as trial run tests before conducting the final summative validation, helping to avoid costly redesigns or repeat validation due to unresolved usability issues or protocol weaknesses.
- Obtaining Institutional Review Board Approval Causing Delays in Submissions: Many companies may believe that formative tests require elaborate institutional review board (IRB) approval regarding participant safety. In most cases, usability testing do not present the risk of participant harm (i.e., no therapy is actually delivered because the concentration is on user interaction with the UI). However, manufacturers may choose to protect themselves from liability by gaining IRB approval, but this can take the form of ‘expediated reviews,’ given the low complexity of the testing compared to the clinical trials.
- Assumptions About Documentation Burden: From an FDA and IEC 62366-1 compliance perspective, formative evaluations do not require extensive documentation in premarket submissions. FDA reviewers merely want to know if the manufacturer has conducted iterative testing on the product and has made the best effort to remove design flaws related to safety. In recent years, however, FDA reviewers have increasingly requested formative evaluation records to demonstrate adequate preparation prior to summative usability validation – a shift from earlier submission expectations.
Simulated Use Testing (Validation)
Summative testing, also referred to as simulated use validation testing, is the primary source of evidence supporting use safety under IEC 62366-1 and the FDA Human Factors guidance. To avoid obvious safety concerns to test participants, summative testing can be conducted under ‘simulated use’ conditions that are representative of real-world conditions.
Summative testing differs from formative evaluations in purpose and executions. It is not meant to be exploratory and does not seek design input; rather, it serves as a final demonstration that the device can be used safely and effectively. Participants are not coached, interrupted, or corrected during task performance.
Use scenarios are designed to reflect typical sequences of interaction with the device and must include all tasks identified as high risk in the use‑related risk analysis. Training and familiarization are provided in a manner consistent with expected real‑world conditions, including consideration of learning retention and potential memory decay. Instructions for use are made available during testing, but participants are not required to review them unless they choose to do so.
User performance is observed and categorized as success, failure, or success with difficulty (e.g., hesitation, self‑correction, or confusion). These observations, along with any failures, are documented for post‑test analysis. Post‑test interviews are conducted to understand the root causes of observed difficulties or errors and to gather participant feedback on task complexity.
Study results must support an overall conclusion regarding use safety. This determination is based not on predefined quantitative success thresholds, but on whether any residual patterns of use‑related problems remain attributable to the UI, labeling, or instructions.
Design and Interpretation of Summative Test Results in Simulated Use Validation Studies
The design, conduct, and interpretation of summative simulated use testing represent one of the most challenging aspects of Human Factors and Usability Engineering (HF/UE). These studies serve as the primary evidence supporting use safety and are distinct from conventional usability evaluations. Their value lies not only in observed task performance, but in the post‑test analysis required to determine root causes of use errors and difficulties.
As described in FDA guidance and IEC 62366-1, summative testing provides manufacturers with a forward-looking assessment of real-world device use. Unexpected or undesired outcomes observed during testing should be investigated using an approach analogous to postmarket adverse event analysis. Thus, observations in these tests that are either unexpected or not desired are investigated in the same manner as an adverse event. The objective is to identify root causes of errors occurring during critical tasks and to determine whether these issues can be mitigated prior to regulatory approval and commercialization.
Submission to the FDA pre-market process has generally required the inclusion of summative validation test results, especially for higher risk devices. Although overall study quality has improved over time, several recuring deficiencies continue to be observed:
- Insufficient Linkage to the User Related Risk Analysis (URRA): Test protocols should be explicitly justified using the URRA (Figure 2), with use scenarios mapped to critical tasks from a risk perspective. Critical tasks must be clearly documented rather than implicitly inferred by the reviewer. Clear presentation of task analysis, risk controls, and validation activities strengthens regulatory review.
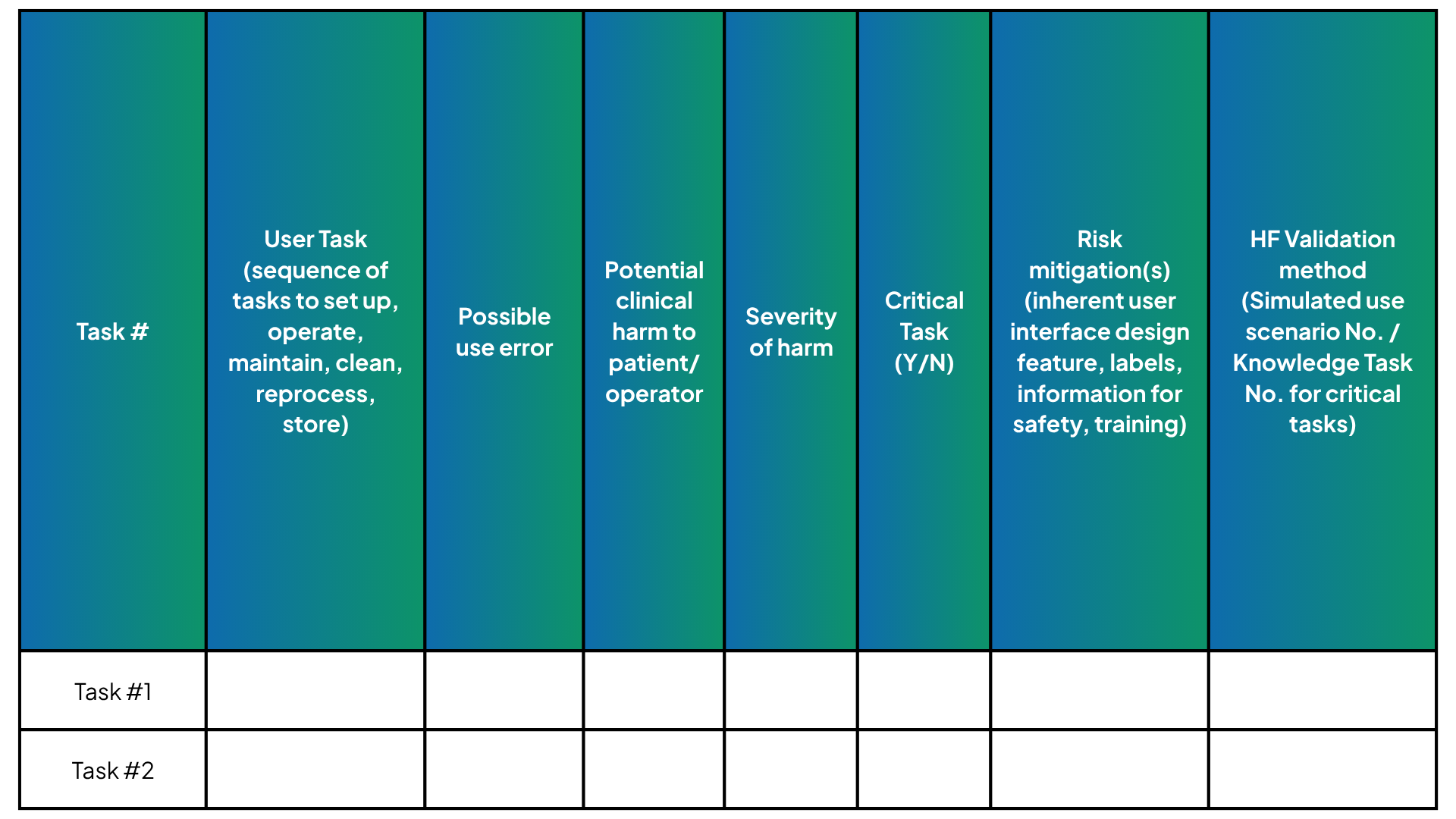
Figure 2: Example of task, risk control, testing scheme
- Reporting Success Rates Without Establishing Use-Safety: Numerical success rates alone are insufficient. Manufacturers must evaluate and discuss failure cases to determine whether even isolated use errors could plausibly lead to serious patient harm. The overall use‑safety argument should be grounded in error analysis, not percentages.
- Overreliance on Subjective Preference Data: Ease‑of‑use ratings and user preferences do not constitute evidence of safe and correct use. Subjective metrics may support conclusions but cannot substitute for objective evaluation of task performance and error mitigation. Believing the device is easy to use does not equate to demonstration of correct use or a cogent argument to regulatory reviewers.
- Inadequate Representation of User Groups or Enough User Groups: Both FDA guidance and IEC 62366-1 recommend including a minimum of 15 participants per identified user group. Determination of appropriate user groups should consider who performs the tasks, relevant user profiles, and differences in training, experience, or physical or cognitive capabilities. Home‑use devices or patient‑use combination products may require additional user groups, such as those with visual or cognitive impairments. Presubmission interactions with the FDA are encouraged to align on testing strategy.
- Lack of Delay Between Training and Testing: Simulated use testing should include a time interval between training and task execution to account for learning decay and reflect real‑world use conditions. In some combination product submissions, the FDA may require a trained and an untrained group.
- Attribute Failures to “Human Error” Without Analysis: Summative test results should be analyzed on a case-by-case related to observed failures and patterns of difficulties exhibited by participants. Merely attributing a failure to ‘participant just forgot’ during execution of a critical tasks is not a solid argument that the interface is safe to use, as the interface should support the user in all ways possible during the task performance. Post-test interviews are critical for understanding user reasoning, points of confusion, and causal factors. Asking participants why they were confused, what led them to believe the device worked a certain way or why they made an error is absolutely essential to the analysis process.
- Failure to Validate Instruction for Use (IFU) When Relied Upon as a Primary Risk Control: When manufacturers identify the IFU as a key risk mitigation measure, regulators typically expect targeted testing to validate its effectiveness. FDA guidance emphasizes that reliance on labeling alone is generally insufficient, and that safety should be designed into the user interface wherever feasible.
Human Factors and Usability Testing as a Competitive Advantage
Beyond compliance, Human Factors and Usability Testing offer a strategic advantage. Devices that are intuitive, efficient, and aligned with real world workflows are more likely to:
- Be adopted by clinicians
- Reduce training burden
- Improve user satisfaction
- Minimize postmarket issues
- Strengthen brand trust
In an increasingly competitive MedTech landscape, usability is not just a regulatory requirement, it is a differentiator.
Conclusion
Human Factors/Usability Engineering has become a vital part of the product development process ensuring medical device ease and safety of use. Worldwide, regulatory organizations require a systematic oversight and review process regarding manufacturer compliance with the usability engineering standard, IEC 62366-1. The FDA pre-market review process now routinely includes human factors user-related risk analysis and validation testing of the device UI with intended users.
HF/UE activities should be conducted throughout all phases of device design and development, including preliminary task and user-related risk analysis, UI design and evaluation, and final summative validation testing in simulated use.
The HF/UE process should be aligned with the overall risk management process for the device. Final decisions regarding use risk acceptability should be based on the results of simulated use testing with representative users evaluating the presence of any remaining patterns and failure and difficulties on tasks with critical risk implications.
Formative and summative Human Factors testing serves distinct but complementary roles in medical device development. When applied strategically, they enable manufacturers—regardless of size—to design safer devices, streamline regulatory pathways, and reduce overall development risk.
- Formative testing provides the insight needed to design effectively
- Summative testing provides the evidence needed to demonstrate safety
Together, they form the foundation of a robust, scalable Human Factors and Usability Testing strategy that supports both compliance and commercial success.
Frequently Asked Questions (FAQs)
Is usability different from human factors?
The term “usability engineering,” often used as a synonym for ‘human factors’, is also focused on creating qualities of UIs that result in rapid learning, user satisfaction, and efficient interaction. The term “usability” is a multi-dimensional quality that refers to the ability of a human to interact easily and relatively error-free with a system or product.
What standards or regulatory guidance should you be aware of for usability of medical devices?
For the US, the FDA recognizes IEC 62366-1, Medical devices – Part 1: Application of usability engineering to medical devices, ANSI AAMI HE75, Human factors engineering – Design of medical devices and FDA guidance, Applying Human Factors and Usability Engineering to Medical Devices, issued February 2016.
The international regulatory community has incorporated IEC 62366-1, Medical devices – Part 1: Application of usability engineering to medical devices, as part of the approval process outside the U.S.
Is Human Factors testing required for all medical devices?
The FDA pre-market review process now routinely includes human factors user-related risk analysis and validation testing of device UI with intended users. This is especially noted for high-risk devices and software as a medical device (SaMD).
How many users are needed for formative testing?
There is no fixed or minimum number of users required for formative evaluations. Both IEC 62366‑1 and the FDA’s guidance Applying Human Factors and Usability Engineering to Medical Devices emphasize that formative testing is iterative and exploratory, with the goal of identifying usability issues and use‑related risks—not statistically validating performance.
While regulators do not mandate a specific sample size, many manufacturers find that 8-10 representative users can reveal 90% of existing design flaws.
How does summative testing differ from other usability testing?
There are several important differences in test methodology regarding summative simulated use tests compared to other usability evaluations such as formative testing. The summative test is not meant to be an exploratory effort seeking inputs on design features and should be performed on the final design of the device. It includes representative users, environments, and use scenarios and follows a structured, predefined protocol which focuses on critical tasks and worst-case conditions. The summative testing will be provided in a formal report for regulatory submission.
Can strong formative testing reduce the risk of summative test failure?
Yes. Iterative formative testing significantly increases the likelihood of successful summative validation and smoother regulatory review.
Resources and References
FDA guidance for Industry and Food and Drug Administration Staff – Applying human factors and usability engineering to medical devices issued February 3, 2016
ANSI AAMI HE75:2009/(R)2018, Human factors engineering – Design of medical devices (FDA recognized #5-57)
IEC 62366-1 Edition 1.1 2020-06 Consolidated Version, Medical devices – Part 1: Application of usability engineering to medical devices (FDA recognized #5-129)
ISO 14971 Third Edition 2019-12, Medical devices – Application of risk management to medical devices (FDA recognized #5-125)
Wiklund, J. Kendler and A. Strochlic, Usability Testing of Medical Devices. CRC Press-Taylor & Francis Group (2011)
