As the COVID-19 Pandemic continues to evolve, the IVD industry has risen to the challenge of producing diagnostic tests to support the public health emergency. This has resulted in an unprecedented amount of new devices entering the market under the Emergency Use Authorization (EUA) pathway. While the EUA pathway is still available, the window for pursuing a EUA is slowly shrinking and being replaced with full U.S. Food and Drug Administration (FDA) approval to remain on market. With clinical studies requiring six to 18 months to complete, it is imperative that IVD manufacturers are proactive to identify gaps, prepare pre-submissions and execute clinical studies.
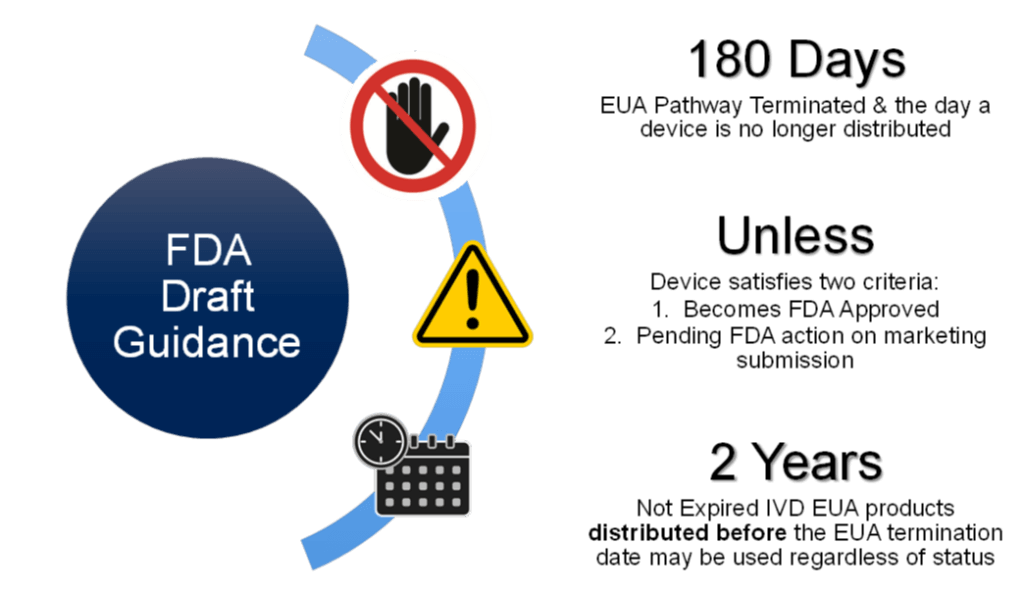
In December 2021, the FDA released two draft guidance documents: 1) “Transition Plan for Medical Devices Issued Emergency Use Authorization (EUA) During the Coronavirus Disease 2019 (COVID-19) Public Health Emergency” and 2) “Transition Plan for Medical Devices That Fall Within Enforcement Policies Issued During the Coronavirus Disease 2019 (COVID-19) Public Health Emergency,” indicating an end to the EUA pathway in 180 days from draft approval.
Although providing advance notice, the process is rigorous when transitioning from EUA to 510(k), in particular for Manufacturers with limited experience with infectious diseases FDA marketing submissions. It is therefore imperative that IVD manufacturers embark on this journey early with FDA collaboration to avoid costly delays and potential loss of sales.
Whether a Manufacturer chooses to complete a formal 510(k) submission or apply for a EUA, NAMSA understands and has extensive experience bringing infectious disease assays to market via traditional pathways (i.e. 510(k), De Novo or PMA) as well as utilizing FDA templates provided for EUA application.
Examples of Support Provided by NAMSA
- Pre-EUA Preparation
- EUA Submissions
- Transition from EUA to de novo and 510(k)
- Notification Pathways
- Complex Development and Manufacturing Plans
- Protocol Development
- Site and Laboratory Identification | Site and Laboratory Qualification
- Monitoring | Data Management | Biostatistics
FDA EUA Template Expertise
- Molecular | Antigen | Serology (+ Neutralizing Antibodies)
- Home Collections for Laboratory Testing
- Tests Intended for POC | Home and OTC use with Considerations for User | User Environment and Usability Testing Obligations
COMPLIMENTARY CONSULTATIONS
NAMSA, as the industry leader in IVD regulatory knowledge and product development, is proud to have partnered with several domestic and overseas IVD Sponsors to help them achieve success. If you are seeking an experienced IVD partner and need assistance with IVD regulatory, clinical, reimbursement or preclinical studies, please reserve your complimentary consultation to learn more: https://namsa.com/namsa-expertise/subject-matter-experts/.

IVD and COVID-19 RESOURCES
We invite you to access the below complimentary resources to learn more about the strategies and best practices that can assist you with addressing COVID-19, IVD product development and IVD regulatory/clinical guidance.
IVD REGULATORY GUIDANCE
Blog Post: FDA Issues COVID-19 Transition Plans for EUA and Enforcement Actions
Blog Post: CDRH COVID-19-Related Workload Impacts Q-Submission Reviews
Blog Post: Diagnostic Developers Look to EUA Submission to Combat COVID-19
Webinar: Transitioning COVID-19 EUA to 510(k) IVD Products: Planning to Execution
IVD CLINICAL GUIDANCE
White Paper: Expanding Your Market: Submitting U.S. EUA COVID-19 Products For EU IVDR Compliance
Webinar: Considerations for Clinical Study Start-Up in a Virtual Environment
White Paper: Navigating Clinical Trials in the Era of COVID-19: An Assessment of Short-Term and Long-Term Strategies
Blog Post: IVD Clinical Trials in the Era of COVID-19: Time to go Virtual?
Webinar: How to Manage Clinical Study Data During the COVID-19 Pandemic
Webinar: The COVID-19 Pandemic – Proper Clinical Study Management & Protecting Data Validity