EU MDR and IVDR Technical Documentation for Medical Devices
The European Medical Device and In-Vitro Regulations (MDR and IVDR) require manufacturers to prepare detailed Technical Documentation to obtain and maintain CE Marking of medical devices. In most cases, this documentation will be reviewed by your European Notified Body and contains a wide array of information about your device: specifications, manufacturing process, design controls, test reports, risk assessments, clinical data, and much more.
Quick Facts about EU Technical Documentation:
- Technical Documentation is the new name for Technical Files and Design Dossiers
- It is required for all classes of medical devices and in-vitro diagnostic medical devices
- Technical documentation includes documentation from post market surveillance too
- MDR/IVDR Annex II and Annex III outline basic documentation requirements
- Some Notified Bodies have issued their own guidance for technical documentation submissions
Gap Assessment and Strategic Advice for Compliance with Current MDR/IVDR Requirements
Many manufacturers seeking CE Marking for devices already have some of the information required. Nonetheless, it can be overwhelming to figure out exactly what is needed under the EU MDR/IVDR, and whether what you have is acceptable to your Notified Body. Our team of consultants includes ex-Notified Body reviewers who can perform a detailed review of your existing documentation. We can identify the gaps and weaknesses, and prepare a plan outlining the procedures, documentation, and testing you need in order to prepare MDR/IVDR compliant technical documentation. If you are transitioning an existing MDD/IVDD-certified device to the EU MDR/IVDR, we can conduct a thorough assessment of your current technical file or design dossier so you know where to focus your efforts prior to your next Notified Body audit. For manufacturers with large product portfolios, we can also provide strategic advice for transitioning your devices to the MDR/IVDR.
Components of Your Technical Documentation
With gaps in your documentation identified, we can help you flesh out what is needed for your technical documentation Typically, we get involved assisting companies with the following issues.
- Device classification and rationale, conformity assessment routes
- Identification of all applicable standards for your device
- Design input, output, review, verification, and validation
- Reviewing current testing reports and recommendations for “state of the art” updates
- Evidence supporting indications and contraindications for use
- Clinical Evaluation Report (CER)/Performance Evaluation Report (PER) gap assessments, preparation and literature reviews
- Risk management assessment and file compilation
- Full package labeling and IFU review for compliance with EU MDR/IVDR
- Reviewing your Post-Market Surveillance (PMS) and post-market clinical follow-up (PMCF) or post-market performance follow-up (PMPF) activities including planning, conducting, and reporting
- Biological safety and CMR/ED materials
- Compliance with aspects of ISO 13485:2016 that feed your technical documentation.
- Specialist support for high-risk devices (e.g. devices incorporating medicines and non-viable human or non-human tissues)
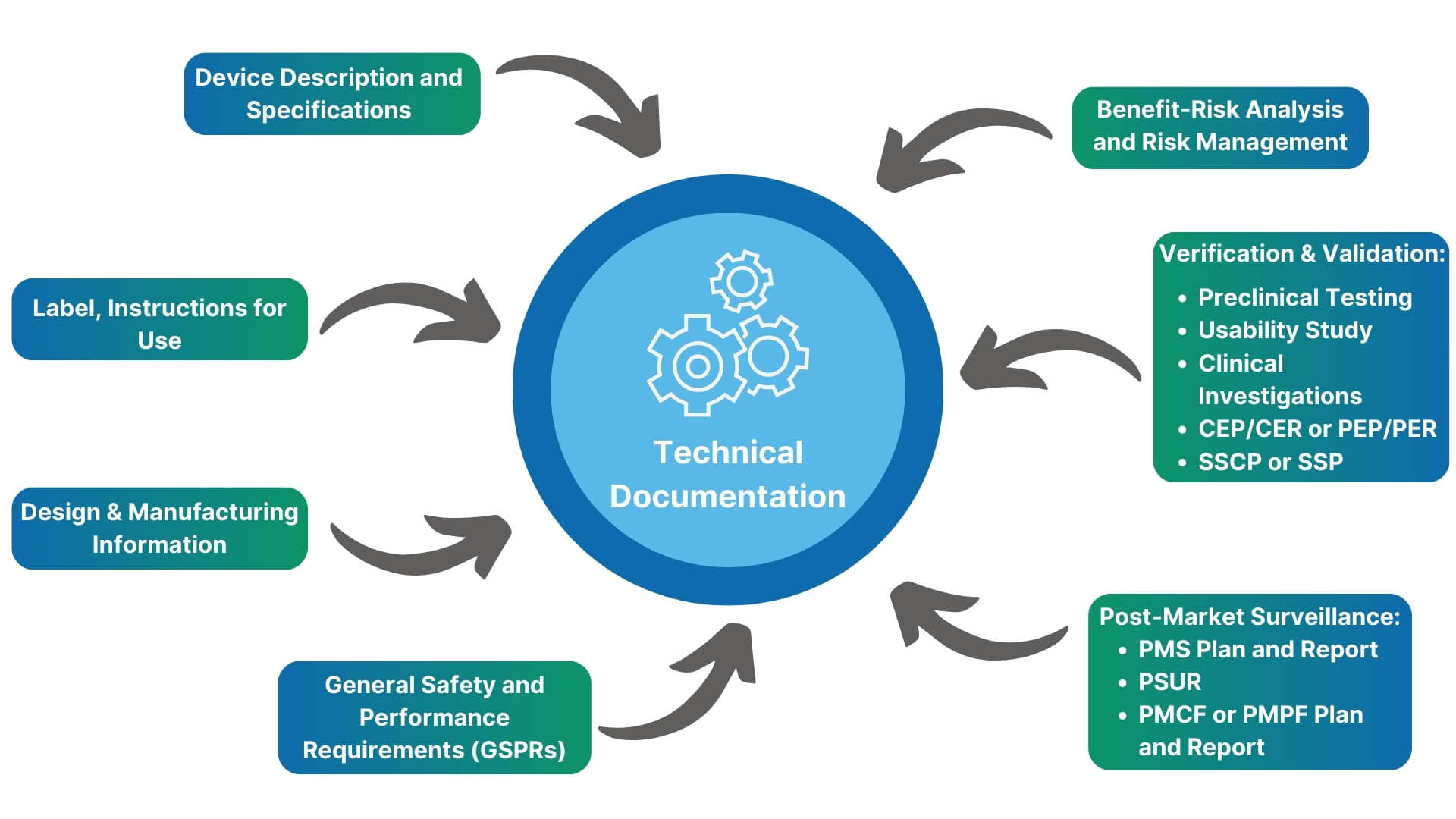
EU Technical Documentation Compilation
Our team prepares MDR/IVDR compliant technical documentation for a wide range of devices and device families. Depending on your needs, we can provide simple compilation of the technical documentation through to writing of all key documents. In addition, there are a suprising number of continous processes connected to your technical documentation including:
- Risk management
- Clinical evaluation/Performance evaluation
- Post-market surveillance
- Post-market clinical follow-up/Post-market performance follow-up
We will take the time to ensure we understand the level of support you need, any particular formatting requirements and time constraints for your project at the outset. We will let you know what documents you need to provide to us and when. We flag any issues as they arise and we can adapt the project if you need additional technical support. Our systematic and proactive approach ensures we create the strongest possible technical documentation and minimize questions and review times when your technical documentation is submitted.
We Know What Your Notified Body Expects to See
After spending so much time and effort gathering clinical evidence, nobody wants to receive major “findings” after a Notified Body review of their technical documentation. NAMSA provides comprehensive range of services to ensure technical documentation meets the MDR/IVDR requirements and the expectations of Notified Bodies. We also offer a much wider range of services than most regulatory consultancies, including preclinical research, testing, and clinical trials. Our unique perspective allows us to spot potential issues in trial data or testing you may have performed previously, and address them before they become big and costly. If you choose to work with NAMSA for the clinical and testing phases of your device development, we can help reduce the risk that your clinical evidence would be rejected by your Notified Body, increasing the chances of a successful submission for CE certification.
Meet Our Team of Regulatory Experts
-
Matt Royle, PhD
 Principal Regulatory ConsultantView Bio
Principal Regulatory ConsultantView Bio -
Kevin Butcher
 Principal Regulatory ConsultantView Bio
Principal Regulatory ConsultantView Bio -
Jane Arnold-Round, MSc
 Senior Principal Consultant, RegulatoryView Bio
Senior Principal Consultant, RegulatoryView Bio -
Rachel Gibbs, BSc, PhD
 Principal Regulatory ConsultantView Bio
Principal Regulatory ConsultantView Bio -
Thomas Miramond, PhD, MSc, M-Eng.
 Senior Regulatory ConsultantView Bio
Senior Regulatory ConsultantView Bio
Request a Meeting with a NAMSA EU Documentation Expert
Commonly Asked Questions about EU Technical Documentation
Technical File was the term used under the previous Medical Devices Directives (MDD 93/42/EEC, AIMDD 90/385/Ee and IVDD 98/79/EC). When the Medical Devices Regulations (2017/745 and 2017/746) were introduced, the terminology was changed to “technical documentation” to highlight the fact that far more supporting information is expected from European regulators than a simple “file.”
If you have a class I device which is non-sterile, not a resusable surgical instrument and does not have a measuring function, you do not need CE certification from a Notifed Body, but you do still need technical documentation. A competent authority may ask to see your technical documentation so it’s still important to keep it available and up to date. Meeting your regulatory and legal responsibilities is an important consideration to avoid business risks, even for self declared products. If you have a custom made device you still need some technical documentation. This needs to comply with Annex III of the MDR.
If you have a Class A device that is non-sterile, you do not need CE certification from a Notified Body, but you do still need technical documentation. A competent authority may ask to see your technical documentation so it’s still important to keep it available and up to date. Meeting your regulatory and legal responsibilities is an important consideration to avoid business risks, even for self-declared products. This needs to comply with Annex II & III of the IVDR.
The good news is that your existing submission may contain much of the data and information needed for CE certification but there are some significant differences. NAMSA can review your documentation, identifiy the gaps, and work with you to prepare technical documentation which will fully meet the requirements of the MDR/IVDR.
Related Services That May Interest You

Clinical Evaluation Reports

PMCF Plans, Surveys and Reports
