Your cart is currently empty!
Medical Device PMCF Plans, Surveys, and Reports
If you market devices in Europe, you already know that your clinical evaluation obligations under the EU Medical Device Regulation don’t end when you get CE Marking. The Medical Device Regulation (MDR) requires you to proactively gather data to assess ongoing device safety and performance. Known as Post Market Clinical Follow-up, PMCF is an important part of your overall Post Market Surveillance (PMS) activities. NAMSA can help you strategize on which data to collect, how to collect it, and prepare a robust PMCF report that meets the expectations of your Notified Body.
Quick facts about Post Market Clinical Follow-up:
- Required in Article 61 and Annex XIV (Part B) of the European MDR (2017/745)
- Critical component of Post Market Surveillance activities
- Applicable to all classes of medical devices, even those with a long and safe performance record
- PMCF report becomes part of your Clinical Evaluation Report (CER)
- Applies throughout entire life cycle of the medical device
- Intensity of PMS proportionate to risk class and type of device
How NAMSA Can Help You Meet PMCF Requirements
Post Market Clinical Follow-up is a systematic process and consists of several phases as seen below.
Step 1: We Can Help You Define Your Strategy
We work with our clients to create a solid PMCF strategy before rushing directly into planning mode. Why? To satisfy MDR requirements (and your Notified Body) it’s important that you collect the right data in the right way. To come up with a strategy, NAMSA will evaluate several factors including costs, practicality, timelines, quantity of marketed devices, scientific rationale and business decisions. We will outline a strategy with input from you and our clinical and regulatory teams. Once the strategy has been agreed upon, we use it to inform decisions on your PMCF plan.
Step 2: Use Your Strategy To Craft A Medical Device PMCF Plan
The PMCF plan is a document which outlines what PMCF activity(s) you plan to carry out for a particular device. MDCG 2020-7 provides a deceptively simple template for creating a PMCF plan. This PMCF template tells you what is needed, but not how to do it or what your Notified Body expects of you. Our in-house clinical and regulatory teams have assisted medical device manufacturers worldwide with Post Market Clinical Follow-up and will develop a thorough PMCF plan that specifies the methods and procedures you will use to collect data and how you will:
- Confirm the ongoing safety and performance of your medical device
- Identify unknown side effects, emergent risks, off-label use or systematic misuse
- Ensure that the benefit-risk ratio outlined in your risk management plan is still acceptable
In most cases, post market clinical studies will not be required if available data is adequate and the risks posed by your device are low. Typically, other activities such as literature reviews and surveys may be sufficient. If that’s the case, we will justify this in your PMCF plan.
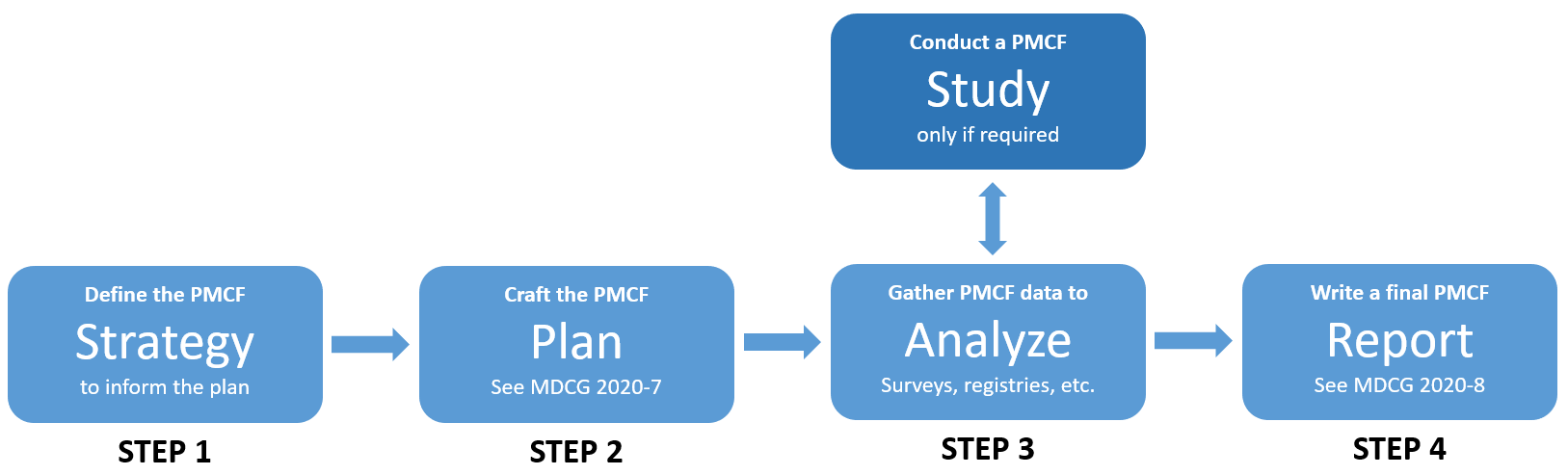
Step 3: Gather Data Via PMCF Surveys, Registries, And More As Specified In Your Plan
Although the EU MDR allows some exceptions, for most devices PMCF data must gathered continually. Post market surveys are one of the most commonly used methods of collecting data from users and patients. NAMSA has deep experience designing, fielding and analyzing PMCF surveys, along with many other sources of post market data as shown below. Our team will help you collect new PMCF data and review existing data you have available from sources including:
- High quality clinical surveys
- Focus groups
- Individual case reports
- Usability surveys including feedback from doctors or patients
- Feedback from sales or account managers
- Published literature on current “state of the art” or equivalent devices
- Clinical registry studies
- Full or partial scope post market clinical studies
Conducting A Medical Device PMCF Study
The depth and breadth of data collected will largely depend on the risk class of your device. If the medium or long-term safety and clinical performance of your device is well documented with real world evidence, a post market clinical study may not be required. However, if a PMCF study is required, NAMSA has teams based in the US and Europe who can plan and manage your study, including protocol development, study management, patient/user recruiting, biostatistics and more.
Step 4: Write The PMCF Evaluation Report
After all data has been collected and evaluated, it’s time to write the report. Once again, there’s a template for that! MDCG 2020-8 provides a basic framework for what should be included. Because NAMSA has worked on so many PMCF projects representing a wide variety of devices, we have a very good sense of what Notified Bodies are looking for. Data from the report will be used to update your clinical evaluation report, risk management file, Summary of Safety and Clinical Performance, etc. Again, because we are one of the largest medical device CROs, we have the in-house expertise to handle all aspects of your PMCF project, from strategy to study to summary reporting.
Request a Meeting with a NAMSA PMCF Expert
Commonly Asked Questions about PMCF
PMCF applies to all medical devices sold in Europe. In a nutshell, PMCF is the ongoing process of updating the clinical evaluation report (CER) that was initially prepared and reviewed before the device was placed on the EU market. Under the EU Medical Device Regulation (2017/745), clinical evaluation does not stop once the device obtains CE Marking – PMCF must be conducted throughout the entire life cycle of the device. The requirement to conduct PMCF is mandated in Article 61 and Part A of Annex XIV of the MDR.
PMCF is always required for all classes of medical devices, even self-certified Class 1 devices. It does not matter if your device has been safely used for decades in Europe – you must proactively and regularly gather evidence that the device continues to be used safely and is effective. The intensity of how you approach PMCF does vary significantly depending on device classification, and your plan for collecting and analyzing data is documented in your PMCF plan.
Circumstances that may justify PMCF studies may include: innovation (new material, new technology, new indication etc..), significant changes to the device or its intended use, invasiveness of the procedure, high-risk anatomical locations, high-risk populations, the severity of the disease treated, high skills necessary to use the device and reproduce the clinical results, unanswered questions about the long-term safety and performance, or identification of a sub-population which may have different risk/benefit ratio.
PMCF studies may also be needed for legacy devices (devices already on the market under the MDD) in order to meet the requirements of the MDR.
Meet Our Team of MDR Regulatory Experts and Medical Writers




Related Services That May Interest You
Global RA/QA Consulting

FSP Outsourcing
