
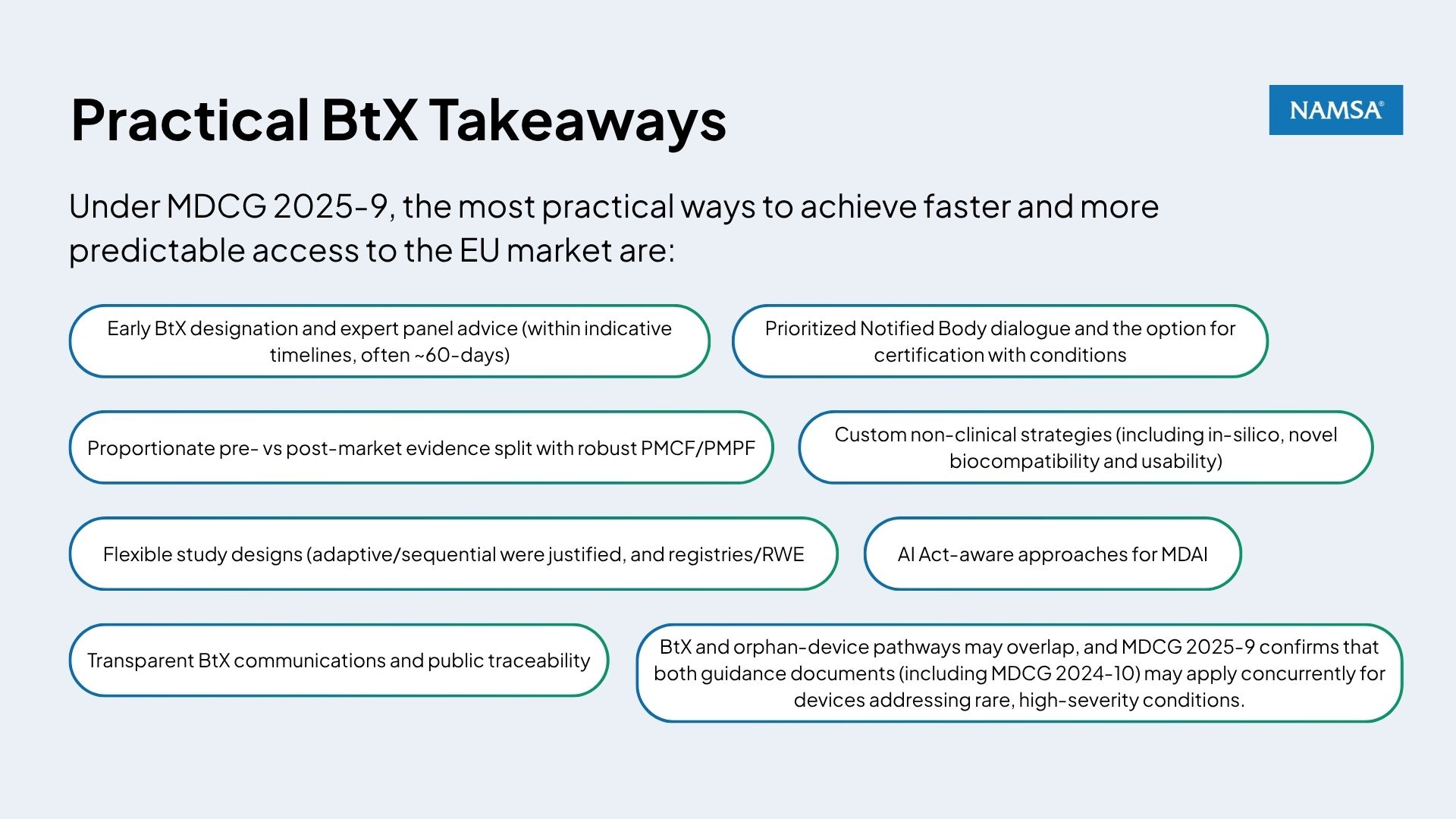
MDCG 2025‑9, “Guidance on Breakthrough Devices (BtX) under Regulations 2017/745 & 2017/746,” creates a practical process for identifying, evidencing, and certifying EU breakthrough medical devices and IVDs, linking early expert engagement, proportionate evidence plans, and prioritised Notified Body review to accelerate safe patient access. For manufacturers, this guidance translates into earlier structured interactions, flexible clinical/performance strategies, and lifecycle evidence plans where some confirmatory clinical data can shift to the post‑market stage with appropriate controls, without lowering MDR/IVDR safety and performance requirements.
Below, we summarise the key requirements and opportunities and outline how NAMSA can help with implementation.
Breakthrough Device Qualification
A device (Medical Device or IVD) is Breakthrough (BtX) if it shows:
- a high degree of novelty in technology, clinical procedure, or application; and
- an expected significant positive clinical impact for life‑threatening or irreversibly debilitating conditions, either versus state‑of‑the‑art alternatives or by fulfilling an unmet medical need.
| Key point: MDCG 2025‑9 emphasises that a device must meet both criteria cumulatively, novelty and significant positive clinical impact, to qualify as a breakthrough device. |
| What This Means in Practice: A strong BtX designation package needs to clearly demonstrate both dimensions across multiple facets of the device — its materials, design, mechanism of action, any AI/ML features, manufacturing approach, intended users, and care setting. Supporting evidence typically draws on laboratory and bench testing, early clinical or performance data, and published scientific literature. Building this package early, ideally at the concept or feasibility stage, positions manufacturers to benefit from structured expert engagement as soon as development allows. |
Early BtX Designation and Expert Engagement, Timelines, and Scope
Manufacturers can request an expert panel opinion on BtX status at any development stage once there’s enough supporting evidence. Panels aim to respond within indicative timelines (often around 60 days) and BtX status can persist through development and after other products enter the market. BtX designation is not automatically invalidated by market entry of competing devices, provided the device continues to meet the novelty and clinical impact criteria.
| Important: BtX status does not confer market exclusivity for that device. |
Beyond their role in CECP and scientific opinions, expert panels can provide early scientific advice, notably for Class III and Class IIb active devices administering or removing a medicinal product under MDR Article 61(2). The Clinical Evaluation Consultation Procedure (CECP) under Article 54 still applies where required, although any prior expert panel advice may be taken into account during that consultation.
Some BtX devices may also qualify as orphan devices under MDCG 2024‑10, and MDCG 2025‑9 confirms that both guidance documents apply in parallel where relevant. Manufacturers should therefore assess BtX eligibility alongside orphan‑device criteria, especially for small‑population, high‑need indications.
| Key Implementation Considerations: Manufacturers will need to plan and author the BtX designation file, structure expert panel scientific advice requests — including the specific questions, supporting background information, and integration with the CECP — and ensure the outputs align with the broader clinical or performance development plan and Notified Body strategy. Regulatory consultants with experience navigating expert panel processes, such as NAMSA’s EU MDR/IVDR team, can help structure these submissions and anticipate the questions panels are likely to raise. |
Evidence Strategy: Balancing Pre‑Market and Post‑Market for BtX
MDCG 2025‑9 confirms that overall clinical/performance evidence requirements do not diminish, but the timing can be rebalanced for BtX when justified (e.g., stronger non‑clinical and short‑/medium‑term clinical data pre‑market, with confirmatory PMCF/PMPF to reduce residual uncertainty post‑market). MDCG 2025‑9 clarifies that although timing may shift, the overall amount of clinical or performance evidence required “does not differ” from non‑BtX devices, the distribution simply becomes more lifecycle‑oriented. Manufacturers must still show acceptable benefit‑risk, a clear PMCF/PMPF plan, and transparent user/patient information about BtX status.
| What This Means in Practice: Rebalancing evidence across the product lifecycle requires careful justification at every stage. Manufacturers should map out pre‑ and post‑market evidence requirements early, define what constitutes sufficient pre‑market data for initial certification, and establish a credible, well‑resourced PMCF/PMPF plan before approaching their Notified Body. This planning is not a formality — Notified Bodies will scrutinise the post‑market commitments closely as a condition of certification. |
Non‑Clinical Foundations for BtX
Because novelty can outpace standards, MDCG emphasises robust preclinical programs, including:
- Targeted literature review to establish the state of the art
- Biological safety tailored to novel materials or nanotechnology
- Bench testing with validated custom protocols and worst‑case conditions
- In‑silico models with demonstrated credibility
- Long‑term simulations with plans to confirm via real‑world data
- End‑to‑end usability from formative to summative testing
Where no harmonised or established standards exist, MDCG requires manufacturers to justify custom test methodologies scientifically and document how each method adequately addresses the relevant risks and performance characteristics.
| Putting This Into Practice : When technologies or materials are genuinely novel, standard test protocols may not exist or may not be directly applicable. Manufacturers need to build scientifically defensible custom verification and validation programs — covering areas such as ISO 10993 biological evaluations, bespoke bench methods, in‑silico modelling strategies, usability engineering, and manufacturing process validation. Documenting the rationale for each custom methodology is as important as the testing itself, and should be built into the technical file from the outset. |
Clinical and Performance Studies: Flexible Designs, Clear Rationale
For medical devices, clinical studies are generally still required for high‑risk products such as Class III and implantable devices. These studies should use an appropriate design, like comparing the new device to current options or using flexible study methods when justified. They may include early “indicator” measures with longer‑term outcomes collected after the device is on the market. The MDCG notes that full randomised controlled trials (RCTs) are often not suitable for many BtX devices, especially when there is no fair comparison option or no appropriate alternative device to test against. In these situations, other well‑designed study approaches, such as adaptive, sequential, or single‑arm studies, may be acceptable if properly justified.
For in vitro diagnostics (IVDs), performance studies should focus on showing the test is scientifically sound and consistently accurate – especially when there are no strong gold‑standard comparators. In situations with limited reference data, carefully planned statistical methods, such as Bayesian approaches or computer‑based simulations, can help strengthen the evidence. For BtIVDs, MDCG notes that challenges in establishing scientific validity may require additional methodologies, including proof‑of‑concept studies, orthogonal methods, or creation of internal reference materials when no comparator assay exists.
| Key Implementation Considerations: Study design for BtX products requires thorough rationale — covering how comparison groups were selected, why the chosen outcomes are meaningful (including patient‑reported measures), and how the design can adapt as results emerge. This spans protocol development, statistical planning, site monitoring, data management, regulatory submissions, and alignment with SSCP/SSP requirements. Manufacturers working with an experienced CRO can benefit from integrated support across these workstreams, helping ensure the study design holds up to both Notified Body and expert panel scrutiny. |
Post‑Market Follow‑Up: Registries, Real-World Evidence (RWE), and Milestones
Where appropriate, manufacturers should aim to prospectively enroll a representative majority of patients or samples exposed to the device within PMCF/PMPF activities, follow them over the long term, and make good use of registries (for devices) or external quality assessment (EQA) programs (for IVDs), along with other real-world data. This helps confirm the product’s safety and performance and spot rare or long-term risks early. Notified Bodies may also grant certificates with specific conditions, such as completing certain post-market milestones and carrying out extra monitoring.
| Putting This Into Practice: Practical post‑market follow‑up plans need to account for registry design and site networks, clear rules for managing emerging evidence, and the milestone reports required for Notified Body oversight and PSURs — all feeding reliably into CER/PER updates. Real‑world evidence programs should be designed prospectively, not retrofitted after certification. NAMSA’s post‑market and clinical teams work with manufacturers to build these programs from the ground up, ensuring they meet Notified Body conditions and produce data that genuinely informs ongoing benefit‑risk assessment. |
Notified Body Interactions: Prioritisation and Structured Dialogue
MDCG expects and encourages Notified Bodies to prioritise BtX devices, ensure early and structured dialogue, and, where needed, seek expert panel scientific advice in parallel with conformity assessment (expert panels aim to respond within indicative timelines (often around 60 days). For SMEs, MDCG reiterates that fee structures and processes must be transparent and proportionate.
Importantly, MDCG 2025‑9 also clarifies that Notified Bodies may issue certificates subject to specific conditions or post-market obligations for BtX devices when pre‑market data are sufficient for initial certification but require post‑market confirmatory evidence to fully characterise long‑term safety, performance, or residual risks. These restrictions may include:
| Condition / Provision | What It Means for Manufacturers |
| Defined PMCF/PMPF activities with mandatory timelines, interim milestones, and specified study designs | Plan post-market studies at the outset; set clear milestones aligned with NB expectations |
| Enhanced surveillance requirements including periodic safety/performance reporting at increased frequency | Budget for more frequent NB reviews proportionate to residual risk |
| Obligations to inform users and patients about BtX status via IFU, labelling, SSCP/SSP | Embed BtX disclosure in labelling and patient-facing documents from day one |
| The MDCG anticipates increased EU-level transparency and potential coordination mechanisms once BtX devices reach the market | Supports EU-level transparency; plan for notification workflow post-certification |
| Non-compliance risk: If a manufacturer fails to meet conditions attached to a BtX certificate, the Notified Body must evaluate the impact on certificate validity — non-compliance may ultimately lead to suspension or withdrawal of the certificate. |
| What This Means in Practice: Manufacturers should prepare for structured Notified Body discussions early, anticipate when CECP consultations may arise, and ensure submissions already address likely conditions — including clear communication materials for users and patients about BtX status. Importantly, the work doesn’t end at certification. Ongoing post‑market monitoring and timely milestone reporting are essential to maintaining certificate validity under a BtX pathway. |
AI‑Enabled Devices (MDAI): MDR/IVDR and AI Act Alignment
High-Risk AI Spotlight
For BtX products that use high‑risk AI, the MDCG highlights several important expectations. Manufacturers need strong data practices, meaning the AI should be trained on reliable, well‑managed, and representative datasets, with active steps taken to reduce bias. They also expect the use of Predetermined Change Control Plans (PCCPs) so that any future AI updates happen in a controlled, predictable, and well‑documented way.
In addition, manufacturers should ensure that their AI systems align with the requirements of the EU Artificial Intelligence Act, which focuses on trustworthy, transparent, and well‑monitored AI, especially for high‑risk medical applications. This includes clear oversight, risk controls, and ongoing monitoring of how the AI behaves in real‑world use.
Finally, there must be continuous evidence that the AI continues to perform well, is reliable across different patient groups and settings, and remains safe over time. All of this should be built into your clinical or performance evaluation, as well as your post‑market monitoring activities.
| Key Implementation Considerations: Developing AI‑aware clinical and performance strategies, robust data governance frameworks, and change‑control processes that satisfy both MDR/IVDR and EU AI Act requirements is a complex undertaking — and the regulatory landscape here is still evolving. Manufacturers should plan for this intersection early, ensuring that AI-specific considerations are embedded in the clinical evaluation, PMS plan, and PCCP from the start rather than addressed as an afterthought. |
Transparency and Public Traceability
Manufacturers should communicate BtX status in labelling/IFU/SSCP/SSP, reference EUDAMED registration, and, where applicable, publicly list clinical/performance study registrations. The MDCG indicates that BtX status should be transparently communicated in documents such as the SSCP/SSP to reflect BtX status and provide clear instructions for users on reporting incidents, complaints, and clinical experience directly to the manufacturer. The Commission aims to develop a public dashboard showing BtX designations and certification status.
| Putting This Into Practice: Transparency obligations under BtX are ongoing, not one-time. CER/PER documents, labelling, and public‑facing materials need to stay current as new evidence becomes available. Manufacturers should build a maintenance schedule for these documents into their post‑market quality system from the outset, treating them as living documents rather than certification deliverables. |
Funding and Parallel Advice Opportunities
The MDCG highlights EU/national innovation funding (e.g., EIC Accelerator, Horizon Europe, EU4Health, InvestEU) and the potential for parallel HTA–Expert Panel Joint Scientific Consultations for certain high‑risk devices. These consultations are particularly valuable for aligning clinical and evidence endpoints early across regulatory and health technology assessment requirements, helping manufacturers avoid costly misalignment between what regulators and payers need to see.
Conclusion
MDCG 2025‑9 doesn’t lower the safety bar. Instead, it explains how companies can organise and justify their evidence for truly new, high‑impact devices so European patients can benefit sooner. This means building a strong foundation of non‑clinical data, doing the right amount of pre‑market clinical or performance testing, and committing to thorough post‑market follow‑up.
With good planning, manufacturers can make use of early expert advice, faster and prioritised Notified Body reviews, and certifications that include clear conditions to speed up access, without compromising safety or performance. NAMSA is ready to support this entire BtX pathway with you, from initial designation through certification and ongoing post‑market confirmation.
Frequently Asked Questions (FAQs)
Does BtX designation mean faster approval or reduced evidence requirements?
No. MDCG 2025-9 is explicit that the overall amount of clinical or performance evidence required “does not differ” from non-BtX devices. What changes is the timing and distribution of evidence generation — with a greater proportion of confirmatory data permissible in the post-market phase through robust PMCF/PMPF plans.
BtX designation does, however, unlock prioritised Notified Body review, structured expert panel engagement, and the possibility of certificates issued with conditions — all of which can reduce delays without reducing rigour.
When in our development program should we apply for BtX status?
Manufacturers can request an expert panel opinion on BtX status at any development stage, provided there is sufficient supporting evidence to demonstrate novelty and expected clinical impact. Early designation — even before a pivotal clinical study — is encouraged, as it allows manufacturers to benefit from structured expert advice when planning their evidence strategy. NAMSA recommends assessing BtX eligibility as early as the concept or feasibility phase so that the designation package and evidence plan can be built in parallel from the outset.
Can a device lose its BtX status if a competitor enters the market?
MDCG 2025-9 clarifies that BtX status can persist through development and after other products enter the market. BtX designation is not automatically invalidated by market entry of competing devices, provided the device continues to meet the novelty and clinical impact criteria. It does not automatically lapse simply because a competing device achieves CE marking. However, BtX status does not confer market exclusivity — a competitor can still market a similar device. Manufacturers should document how their device continues to meet the novelty and clinical impact criteria as the competitive landscape evolves, particularly at each regulatory submission milestone.
How does MDCG 2025-9 interact with the EU AI Act for AI-enabled medical devices?
For BtX devices that incorporate high-risk AI, MDCG 2025-9 highlights the expectation that manufacturers align with EU AI Act requirements — particularly transparency, oversight, bias mitigation, and continuous performance monitoring. Predetermined Change Control Plans (PCCPs) are expected so that AI updates occur in a controlled, documented manner. These requirements are additive to MDR/IVDR conformity obligations and must be reflected in both the clinical/performance evaluation and the post-market surveillance plan.
What happens if we fail to meet the post-market conditions attached to our BtX certificate?
MDCG 2025-9 is clear on this point: if a manufacturer fails to meet conditions or provisions attached to a BtX certificate, the Notified Body must evaluate the impact on the certificate’s validity. Non-compliance may ultimately lead to suspension or withdrawal of the CE certificate.
This makes robust planning of post-market activities — including registry design, milestone reporting, and ongoing NB engagement — critical from day one of certification.
