
The Commission proposal COM(2025) 1023, published 12 December 2025, introduces targeted amendments that will impact MDR and IVDR technical documentation (TD) (Annex II/III) content, the depth of notified body (NB) review, and lifecycle maintenance obligations. The overall intent of the proposal is to remove repetitive “documentation layers”, improve predictability of assessment effort, and introduce specific pathways for mature technologies and niche innovations, without changing the fundamental requirement to demonstrate conformity with the General Safety and Performance Requirements (GSPRs).
Some of the notable changes presented in the commissions proposal are provided below.
1) Technical Documentation Sampling: One Representative Device Per Group
Where Class IIa and non‑implantable Class IIb devices are assessed on a sampling basis, the proposal clarifies that technical documentation assessment is needed only for one representative device per category of devices or generic device group. This is a material simplification for portfolio certification strategies and reduces duplicative file reviews during initial assessment.
In addition, for IVDs, Class B and Class C devices will follow a similar sampling model, in which technical documentation assessment applies to only one representative device per generic device group or category, or, under certain conditions, to the entire portfolio, thereby reducing redundant Technical Documentation reviews across broad IVD families. Class A sterile IVDs will no longer require notified body involvement, eliminating Technical Documentation assessment entirely for this group.
| CURRENT | PROPOSED |
|---|---|
| Full Technical Documentation assessment required for each device within a sampled group; duplicative reviews across broad portfolios. | One representative device per category/generic device group is sufficient. Class B and Class C IVDs follow the same model; Class A sterile IVDs no longer require NB involvement at all. |
2) Well‑Established Technology (WET): Updated Criteria and Reduced Documentation Requirements
The proposal codifies well‑established technology device via a legal definition (simple, stable design; no historical safety issues; well‑known clinical performance; long EU market history) in compliance with MDCG 2020-6, replacing prior list-based logic and enabling proportionality whenever the criteria are met, rather than only for predefined device subsets for new and legacy devices.
Consequently, such devices will not require generation of an SSCP (Summary of Safety and Clinical Performance) (Article 32). An SSCP is required for Class IIb implantable and Class III devices excluding WET devices (and excluding custom‑made/investigational devices). This removes a significant recurring deliverable for eligible mature technologies. Also, the implant card (and leaflets) exemption applies to all WET devices according to this extended definition.
Parallel IVDR simplification: The Performance Evaluation Consultation Procedure (PECP) is proposed for removal, simplifying the Technical Documentation package for Class D IVDs by reducing the volume of material previously required for expert panel scrutiny.
3) Clinical Evidence Packaging: Expanded Recognition of Non‑clinical Evidence
The amended clinical evaluation provisions enable the possibility to justify conformity based on non‑clinical testing methods alone when clinical confirmation is not deemed appropriate – subject to robust justification grounded in risk management and device-body interaction. Examples of non-clinical testing include:
- Bench testing
- In vitro, ex vivo, in silico testing
- Computational modelling/simulation
- Preclinical evaluation
This supports a more technically coherent TD evidence map for device groups with limited feasible clinical data generation.
For IVDs, the proposal introduces similar proportionality in performance‑evaluation documentation by allowing routine blood‑draw studies to proceed without prior authorization and removing the notification requirement for companion‑diagnostic performance studies using leftover specimens. This reduces the volume of study protocols, ethics submissions and related performance‑study documentation included or referenced in the Technical Documentation.
4) Simplified Equivalence: No Competitor Contract Required
The proposal removes the requirement for device manufacturers to have a contract with the manufacturer of an equivalent device when granting access to its technical documentation. This addresses an obstacle that has made equivalence difficult to implement. The focus shifts from contractual access to scientific substantiation and transparency of the equivalence rationale within the Clinical Evaluation Report (CER) and supporting evidence set.
| CURRENT | PROPOSED |
|---|---|
| Manufacturers must hold a formal contract with the equivalent device’s manufacturer granting access to their Technical Documentation — often impossible between competitors. | Focus shifts to scientific substantiation and transparency of the equivalence rationale within the CER and supporting evidence set. No contractual access requirement. |
5) Expert Panel Consultation Expanded to All Class IIb and Class III Devices
Manufacturers of all Class IIb (not only rule 12) and Class III devices may consult expert panels prior to clinical investigation/evaluation to obtain advice on clinical development strategy; both manufacturer and NB are required to consider that advice and justify deviations in the relevant reports. This introduces an EU-level mechanism intended to reduce iterative NB deficiency cycles and increase predictability of TD clinical content expectations.
6) Clinical Investigation Expectation Concentrated on Implantable Class IIb and Class III
The proposal maintains the explicit clinical investigation expectation principally for implantable Class IIb (not IIa anymore) and Class III devices (subject to defined exemptions and proportionality tools, including WET-related provisions where applicable). This supports clearer TD planning boundaries for classes outside that scope.
Planning impact: Manufacturers of non-implantable Class IIb devices should reassess their clinical evidence strategy in light of this change — the current expectation for clinical investigations may be relaxed, opening pathways previously considered unavailable.
7) CECP Narrowed: Class III Implantable Devices Only
The Clinical Evaluation Consultation Procedure (CECP) is limited to Class III implantable devices (with the possibility to add device types by delegated act on safety grounds). This reduces mandatory external scrutiny steps for device types removed from the scope of CECP and therefore reduces associated Technical Documentation “consultation package” overhead.
| Device Type | CECP Status — Current | CECP Status — Proposed |
|---|---|---|
| Class III implantable | Required | Required |
| Class IIb (Rule 12 — administer/remove medicinal product) | Required | Removed |
| Other Class IIb (non-implantable) | Not required | Not required |
| Class D IVDs (PECP) | Required | Removed (PECP eliminated) |
8) Lifecycle Documentation: PSUR Periodicity Reduced
PSUR (Periodic Safety Update Report) update frequency is reduced: Class IIb and Class III PSUR updates occur in the first year after the certificate is issued and every two years thereafter (or upon significant benefit-risk change), while Class IIa PSUR updates are required when necessary (not every two years). This materially reduces routine TD maintenance burden while retaining signal-driven updates.
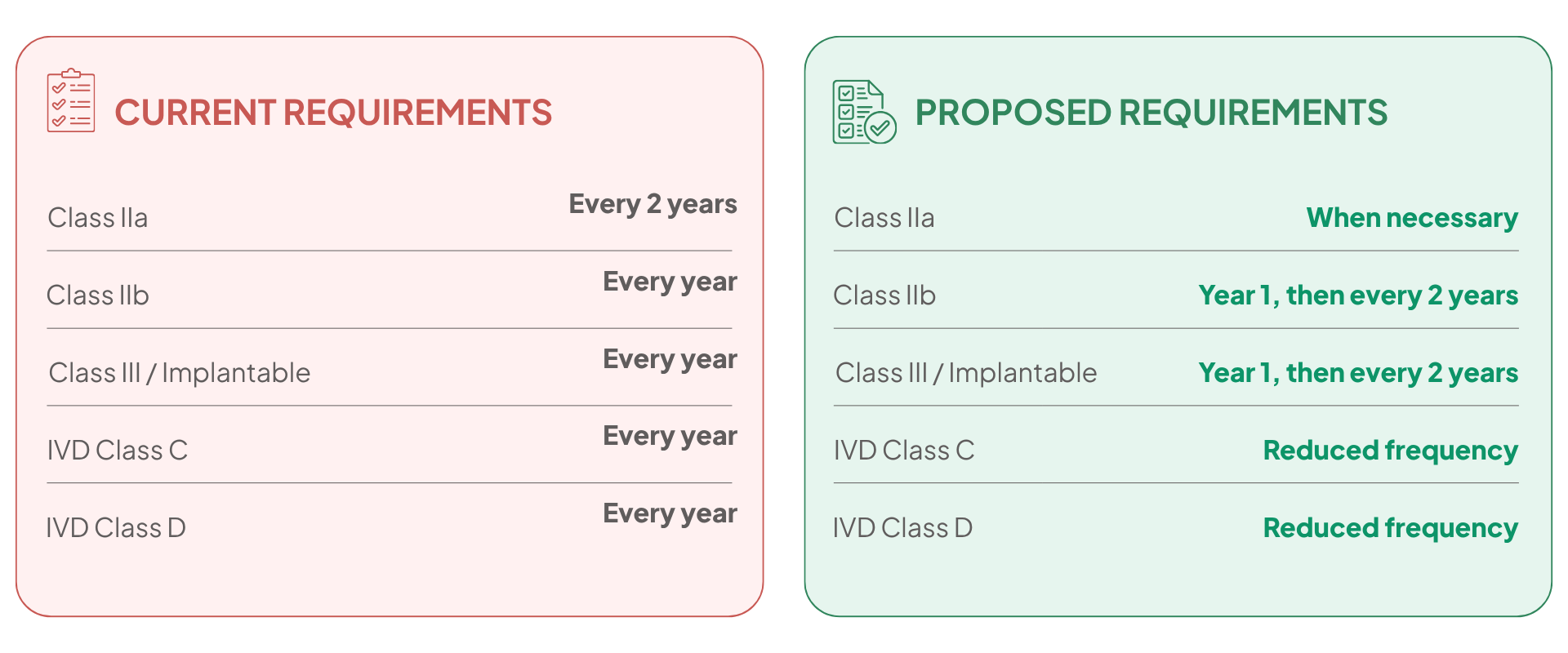
In addition, the proposal reduces duplication of post‑market documentation by enabling integration of certain post‑market clinical follow-up (PMCF) outputs into lifecycle clinical evaluation updates (i.e., fewer standalone report artefacts), while maintaining PMS (post-market surveillance) system obligations.
A mirrored change affects IVDR lifecycle documentation: the reduced frequency of PSUR updates for Class C and Class D IVDs lowers the number of periodic updates that must be integrated into Annex III technical documentation and reduces repeated NB review cycles of performance‑related data.
9) Vigilance and Cybersecurity: Extended Timelines and New Reporting Obligations
For serious incidents not linked to public health threats, death, or serious deterioration, the reporting timeline is extended to 30 days.
New Cybersecurity Obligation: A Dedicated Reporting Channel for Actively Exploited Vulnerabilities
For actively exploited vulnerabilities and severe incidents, with reporting via the MDR electronic system and availability to national CSIRTs and ENISA, requiring corresponding updates to PMS/vigilance procedures and interfaces referenced within TD/QMS documentation.
These cybersecurity reporting obligations apply equally to IVDs and therefore require inclusion of new vulnerability‑monitoring processes and reporting pathways in the Technical Documentation’s PMS and vigilance subsections (Annex III), ensuring manufacturers document the handling of actively exploited vulnerabilities and severe cybersecurity incidents.
10) Breakthrough and Orphan Devices: Tailored Conformity Assessment Pathways
To support innovation and availability for small patient groups, the proposal introduces criteria and a tailored process for breakthrough devices and orphan devices, including expert panel involvement and prioritization/rolling review by Notified Bodies. This is intended to adapt conformity assessment to the specific evidentiary and feasibility constraints of these products and should be reflected in TD planning (modular evidence delivery, staged clinical commitments, defined post‑market data obligations where needed).
Under the IVDR, orphan IVDs may also qualify for continued market availability and prioritized assessment, meaning that Technical Documentation for such devices may follow a staged or modular submission approach with defined post‑market data commitments, similar to the MDR breakthrough/orphan device pathway. This enables proportionate Technical Documentation content for low‑volume, high‑clinical‑need IVDs.
Note: This section of COM(2025) 1023 directly reinforces and expands on the dedicated guidance in MDCG 2024-10 and MDCG 2025-9.
11) Certificate Validity: No Default Time Limit Under Proposed Changes
The proposal removes the default maximum validity period of certificates: certificates are not limited in time unless the NB justifies a limited validity on specific grounds and are maintained through risk‑proportionate periodic reviews rather than a formal recertification cycle.
From a global regulatory architecture perspective, this shifts MDR closer to lifecycle compliance oversight models where market authorization is not routinely re-issued on a fixed multi‑year cadence, but maintained through ongoing post‑market controls and quality system oversight. For example, FDA device market access pathways are risk-based (e.g., 510(k), De Novo, PMA) and are embedded within broader lifecycle controls; manufacturers are subject to quality system requirements under 21 CFR Part 820, and PMA devices have periodic reporting obligations (e.g., annual reports) as part of post‑approval controls.
12) Combined Studies: Single Submission and Coordinated Assessment
For combined studies involving medicinal products, the proposal enables a single application and coordinated assessment approach aligned with the clinical trials framework and related legislative changes (Biotech Act alignment). This reduces parallel submission burdens and supports a more integrated dossier strategy across regulatory regimes.
For combined studies involving medicinal products and IVDs, the ability to submit a single coordinated application similarly reduces duplicated performance‑study documentation within Technical Documentation and eliminates the need for parallel dossier packages across regulatory regimes.
13) Shorter Arbitration Time
The proposal formalizes the “Helsinki procedure” for coordinated Member State decisions on borderline qualification and device classification, with expert panel input where there is substantiated disagreement, and it assigns national notified‑body authorities an ombudsperson role to resolve manufacturer-NB disputes, with a 90‑day handling target.
The formalization of the Helsinki procedure also applies to IVD borderline and classification decisions, ensuring that the Technical Documentation structure and classification‑linked evidence expectations for IVDs are more predictable and harmonized across Member States. This reduces the risk of divergent Technical Documentation requirements or rework caused by inconsistent national classification outcomes.
Related Reading – Published December 2025
MDR: Aspirations vs. Reality in Notified Body Practice
COM(2025) 1023 is a direct response to the tensions exposed by the TEAM-NB Code of Conduct (Version 5.1) and the 12th Notified Body Survey. NAMSA’s in-depth analysis reveals the gap between NB harmonization goals and operational reality — including inconsistent WET handling, application refusal patterns, and certification bottlenecks — providing essential context for understanding why the Commission’s proposed simplifications are needed now.
Practical TD Takeaways
Under the major MDR update proposed in COM(2025) 1023, the most tangible TD simplifications arise from the following:
- Clarified “one representative device” sampling for Class IIa and non‑implantable Class IIb
- Exclusion of SSCP and implant card for WET devices and broader WET applicability via criteria in alignment with MDCG 2020-6 criteria
- Reduced PSUR update cycles
- Removal of the equivalence contract barrier
- Narrowed CECP scope
- Replacement of fixed re‑certification cycles with continuous certificate validity plus proportionate periodic reviews, while adding a new cybersecurity reporting channel to CSIRTs/ENISA and introducing dedicated conformity assessment adaptations for breakthrough and orphan devices
Conclusion
This proposal represents a structural amendment to the MDR/IVDR framework rather than a marginal adjustment: it re‑balances conformity assessment, clinical and performance evidence expectations, post‑market obligations, and EU‑level coordination mechanisms with the explicit objectives of simplification, burden reduction, and improved predictability/cost‑efficiency, while maintaining a high level of health protection.
In practice, it will require implementation time and procedural re‑alignment across the ecosystem. Notified Bodies will need to adapt sampling strategies, surveillance models and operational practices under both MDR and IVDR; competent authorities will need to operationalize the reinforced coordination and dispute pathways; and manufacturers will need to update technical documentation architectures, PMS/vigilance interfaces and clinical and performance evidence development plans to reflect the amended legal requirements in both MDR and IVDR.
If implemented consistently, these measures are expected to support a more predictable time‑to‑market, reduce unnecessary rework, and improve availability and accessibility of safe medical technologies and in vitro diagnostics, thereby enabling better access for EU patients and supporting innovation, particularly for SMEs, breakthrough technologies and niche/orphan indications across both medical devices and IVDs.
Frequently Asked Questions (FAQs)
Does COM(2025) 1023 change the GSPRs or the fundamental safety and performance requirements?
No. The proposal does not change the underlying General Safety and Performance Requirements (GSPRs) or lower the level of evidence required to demonstrate conformity. However, it introduces targeted simplifications and proportionality measures that affect how evidence is generated, structured, and assessed across the product lifecycle.
The intent is to reduce repetitive “documentation layers” and improve predictability of assessment effort — not to lower the standard of evidence manufacturers must ultimately provide to demonstrate conformity. What changes is how and when that evidence is packaged, reviewed, and maintained — not the underlying safety and performance bar.
How does the WET (well-established technology) change affect our existing SSCP obligations?
Under the proposal, devices that meet the WET criteria — including a simple and stable design, well-characterized clinical performance, absence of significant safety concerns, and a sufficiently long and well-documented history of use within the EU — will be exempt from the SSCP requirement under Article 32. This means manufacturers of eligible WET devices would no longer need to generate, maintain, and submit SSCPs to EUDAMED.
Manufacturers must formally assess and justify whether their device meets the WET criteria, and this justification will be subject to Notified Body review as part of the conformity assessment.
NAMSA recommends proactively conducting a WET eligibility review for relevant devices in your portfolio, so you are ready to adjust your documentation program as soon as the legislation is finalized.
What does the removal of the equivalence contract requirement mean in practice for our CER?
Under the current MDR, demonstrating equivalence to a competitor device typically requires a formal contract granting access to the equivalent device manufacturer’s Technical Documentation — often practically impossible between competing manufacturers. This has been a significant obstacle to using equivalence as the clinical evidence route.
Under COM(2025) 1023, the focus shifts to the scientific substantiation of the equivalence claim within the CER itself — manufacturers must provide a transparent, well-documented equivalence rationale based on clinical, biological, and technical characteristics. While this removes the contractual barrier, it does not reduce the scientific rigour expected: a robust equivalence argument will still need to be defensible to the Notified Body. Our CER best practices article provides further context on equivalence documentation.
In practice, while the contractual barrier is removed, manufacturers must still ensure that sufficient technical, biological, and clinical data are available to substantiate equivalence, which may remain challenging in the absence of access to proprietary data.
Does the new cybersecurity reporting obligation apply to all medical devices or only software-based ones?
The cybersecurity reporting obligation introduced by COM(2025) 1023 applies to devices with digital or connected components where actively exploited vulnerabilities or severe cybersecurity incidents may occur.
This means manufacturers of connected hardware, combination devices, and IVDs with digital interfaces all need to assess whether their PMS/vigilance procedures and Technical Documentation (Annex III) reflect the new CSIRT/ENISA reporting pathways. NAMSA recommends a proactive cybersecurity documentation review for all connected and software-enabled devices in your portfolio. Routine cybersecurity risks remain managed within existing PMS and risk management systems; the new obligation specifically introduces structured reporting pathways for high-severity or actively exploited events.
