
Inhaltsverzeichnis
- Warum Human Factors und Usability-Tests wichtig sind
- Einbeziehung von HF/UE in die Medizinprodukteverordnung
- Benutzeroberflächendesign/-bewertung
- Angemessene formative Evaluationen durchführen
- Simulationstests (Validierung)
- Gestaltung und Interpretation summativer Testergebnisse in Validierungsstudien mit simulierter Nutzung
- Menschliche Faktoren und Usability-Tests als Wettbewerbsvorteil
- Fazit
- Häufig gestellte Fragen (FAQs)
- Ressourcen und Referenzen
Usability Engineering, oft synonym mit Human Factors verwendet, konzentriert sich auf die Gestaltung von Benutzeroberflächen (UIs), die eine effiziente und intuitive Interaktion, schnelles Lernen und hohe Benutzerzufriedenheit ermöglichen. Usability ist ein mehrdimensionales Konzept, das beschreibt, wie einfach und zuverlässig Benutzer mit einem System oder Produkt interagieren können, um die beabsichtigten Ergebnisse zu erzielen. Human Factors (HF) und Usability Engineering für Medizinprodukte zielen darauf ab, anwendungsbedingte Risiken und Fehler zu minimieren, um die Sicherheit und Wirksamkeit des Produkts zu gewährleisten. Usability-Tests sind in der Entwicklung von Medizinprodukten nicht mehr optional. Sie tragen entscheidend zur Patientensicherheit, zur Einhaltung regulatorischer Vorgaben und zum wirtschaftlichen Erfolg bei. Zulassungsbehörden weltweit erwarten zunehmend von Herstellern den Nachweis, dass ihre Produkte von den vorgesehenen Benutzern in realen Umgebungen sicher, effektiv und intuitiv eingesetzt werden können.
Trotz dieser Erwartungen stehen viele Hersteller von Medizinprodukten, insbesondere Startups und aufstrebende Unternehmen, vor der Herausforderung, Aktivitäten im Bereich der menschlichen Faktoren effizient und nachhaltig in den Entwicklungszyklus zu integrieren.
Das Verständnis von Zweck, Zeitpunkt und regulatorischem Wert von Human-Factors-Tests ist unerlässlich für die Entwicklung einer konformen, skalierbaren und kosteneffektiven Strategie.
Warum Human Factors und Usability-Tests wichtig sind
Unerwünschte Ereignisse der letzten Jahrzehnte haben besorgniserregende Trends nach der Markteinführung aufgezeigt, die auf Designmängel der Benutzeroberfläche (UI) von Medizinprodukten zurückzuführen sind. Infusionspumpen, automatische elektronische Defibrillatoren, Beatmungsgeräte und Kombinationsprodukte wie Medikamenten-Autoinjektoren weisen in der Vergangenheit häufig anwendungsbedingte Designprobleme auf, die zu Überdosierungen, fehlerhafter Verabreichung, Fehldiagnosen und gefährlichen Therapieverzögerungen geführt haben. Im Rahmen der systematischen Bemühungen der Aufsichtsbehörden zur Fehlerreduzierung wurden Medizinproduktehersteller in den USA und der EU mit den Disziplinen Human Factors und Usability Engineering (HF/UE) vertraut gemacht. HF/UE wird in der Automobil-, Luft- und Raumfahrt- sowie Telekommunikationsindustrie seit über 80 Jahren angewendet, wurde aber erst in den 2000er-Jahren formal in die Medizinprodukteindustrie integriert, mit einem starken regulatorischen Impuls der FDA im Jahr 2011 und einer endgültigen, strengen Durchsetzung ab etwa 2016.
Die Berücksichtigung menschlicher Faktoren und Usability-Tests ist von entscheidender Bedeutung, da sie die Sicherheit, Wirksamkeit und intuitive Bedienbarkeit von Produkten – insbesondere von Medizinprodukten und Software – durch die Bewertung der tatsächlichen Interaktion von Anwendern gewährleistet. Die Ergonomie konzentriert sich auf die Interaktion zwischen Nutzern, Geräten und Umgebungen. Benutzerfreundlichkeit hat einen erheblichen Einfluss auf das Gesundheitswesen, insbesondere hinsichtlich der Gesamteffektivität von Medizinprodukten. Vereinfacht gesagt: Mangelnde Benutzerfreundlichkeit kann die Ausführung von Benutzeraufgaben langsamer und fehleranfälliger sein. Dies beeinträchtigt die Therapie und kann die Patientensicherheit gefährden. Es ist bekannt, dass benutzerfreundliche Produkte beliebter sind und dadurch Marktdifferenzierung und Wettbewerbsvorteile ermöglichen. Daher kann Benutzerfreundlichkeit neben der Risikominimierung auch aus Geschäfts- und Vertriebssicht ein positives Merkmal darstellen.
Einbeziehung von HF/UE in die Medizinprodukteverordnung
Als Reaktion auf die zunehmende Zahl anwendungsbedingter unerwünschter Ereignisse im Zusammenhang mit der Konstruktion von Ultraschallgeräten hat die US-amerikanische Arzneimittelbehörde (FDA) die Überprüfung von Ultraschallgeräten und Ultraschallgeräten als routinemäßigen Bestandteil des Zulassungsverfahrens vor dem Inverkehrbringen integriert. Innerhalb des Center for Devices and Radiological Health (CDRH) werden diese Überprüfungen vom Office of Product Evaluation and Quality (OPEQ) durchgeführt und orientieren sich an den veröffentlichten Leitlinien der FDA. Anwendung von Human Factors und Usability Engineering auf medizinische Geräte.
Ebenso haben internationale Regulierungsbehörden die IEC 62366-1 übernommen. Medizinprodukte - Anwendung von Usability Engineering auf MedizinprodukteAls Teil ihrer regulatorischen Rahmenbedingungen außerhalb der Vereinigten Staaten. Sowohl die FDA-Richtlinien als auch IEC 62366-1 definieren einen strukturierten Usability-Engineering-Prozess, der den gesamten Entwicklungszyklus des Geräts umfasst und in Usability-Validierungstests des finalen Benutzeroberflächendesigns unter simulierten Nutzungsbedingungen gipfelt.
Grundlegende Aktivitäten im HF/UE-Prozess und Abstimmung mit der Geräterisikobewertung
Prozesse der Mensch-Computer-Interaktion und Usability-Entwicklung umfassen die Definition von Nutzergruppen, die Durchführung von Aufgabenanalysen, formative Evaluationen und summative Usability-Validierungstests, um eine sichere und effektive Gerätenutzung zu gewährleisten. Diese Aktivitäten stehen in direktem Zusammenhang mit dem in ISO 14971 definierten Risikomanagementprozess für Medizinprodukte, indem sie die Identifizierung, Minderung und Validierung von Kontrollmaßnahmen für kritische nutzerbezogene Gefahren, wie sie beispielsweise in einer nutzungsbezogenen Risikoanalyse (URRA) dokumentiert sind, beinhalten.
Größte Herausforderungen für Gerätehersteller
Folgende Herausforderungen bestehen weiterhin bei der Umsetzung der Ziele der FDA-Leitlinien und der IEC 62366-1:
- Durchführung angemessener formativen Evaluierungen vor der endgültigen Designvalidierung
- Durchführung und Dokumentation umfassender Nutzungsrisikoanalysen
- Die Konzeption und Interpretation von summativen Testergebnissen im Rahmen von Validierungsstudien mit simulierter Anwendung.
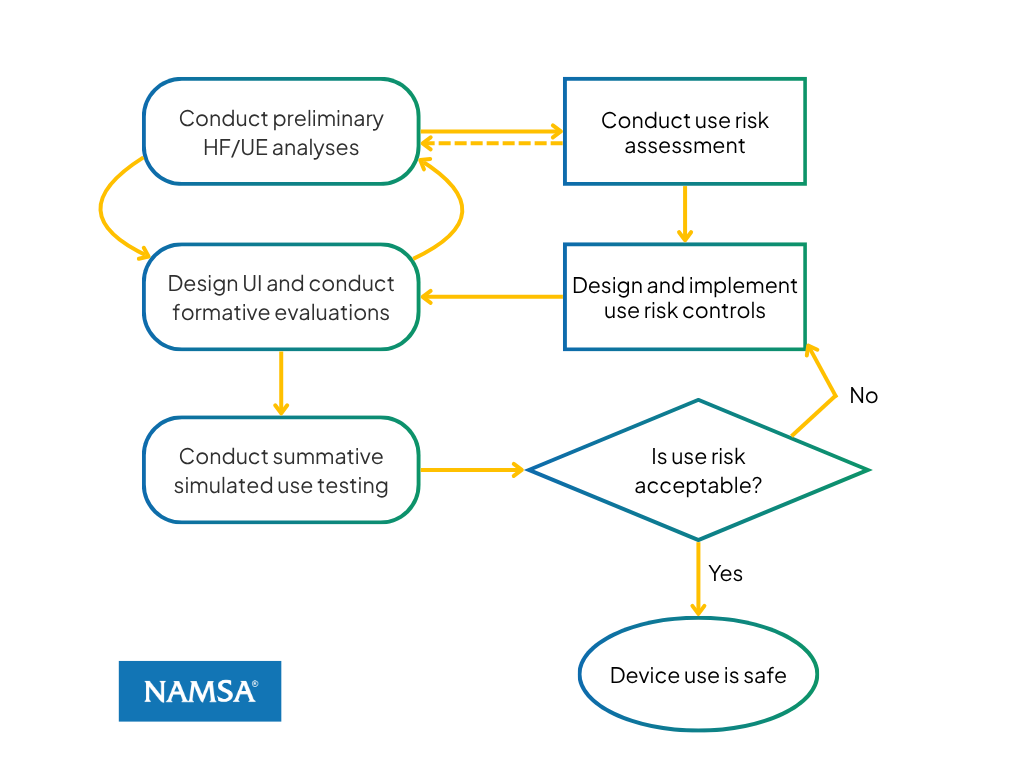
Abbildung 1: Zusammenhang zwischen dem HF/UE-Prozessablauf und dem Risikokontrollprozess nach ISO 14971
Abbildung 1 veranschaulicht den Zusammenhang zwischen den Hauptphasen der Human Factors/User Experience (HF/UE) und den entsprechenden Stufen der Geräterisikobewertung und des Risikomanagements. Links sind die Phasen der HF/UE-Aktivitäten dargestellt, während rechts die Stufen der Risikobewertung und des Risikomanagements gezeigt werden, um die Wechselwirkungen dieser Prozesse aufzuzeigen. Der Zusammenhang zwischen dem HF/UE-Prozess und ISO 14971 lässt sich wie folgt zusammenfassen:
- RisikobewertungDie Risikoanalyse nach ISO 14971 wird durch frühzeitige HF/UE-Aktivitäten unterstützt, die sich durch eine vorläufige Analyse auszeichnen, die sich auf das Verständnis der Benutzer, ihrer Nutzungsumgebung, der Interaktionsaufgaben und potenzieller nutzungsbedingter Gefahren im Zusammenhang mit der Geräteschnittstelle konzentriert.
- Umsetzung des RisikomanagementsDie Umsetzung von Risikokontrollen entspricht der Gestaltung und Bewertung der Benutzeroberfläche, einschließlich des Testens von Schnittstellenfunktionen, die dazu dienen, identifizierte nutzungsbezogene Risiken zu mindern.
- RestrisikobewertungDie Akzeptanz von Risikokontrollen wird durch simulierte Nutzungstests geprüft, die bewerten, ob Restmuster von Nutzungsfehlern bei kritischen Aufgaben weiterhin bestehen und ob nutzungsbedingte Risiken auf ein akzeptabel niedriges Niveau reduziert wurden.
Durch diese Abstimmung wird sichergestellt, dass HF/UE-Aktivitäten vollständig in den Gesamtrahmen für das Geräte-Risikomanagement integriert werden und objektive Nachweise zur Unterstützung der Anwendungssicherheit liefern.
Benutzeroberflächendesign/-bewertung
Sowohl die FDA als auch die IEC 62366-1 betonen einen UI-Designprozess (User Interface), der auf iterativen, formativen Evaluierungen basiert. Diese Evaluierungen werden frühzeitig und während der gesamten Designentwicklung durchgeführt, um die Benutzerfreundlichkeit zu verbessern und nutzungsbedingte Risiken zu minimieren. Formative Evaluierungen verfolgen zwei Hauptziele: die intuitive Bedienung des Geräts zu optimieren und potenzielle nutzungsbedingte Gefahren zu identifizieren und zu beheben. In manchen Fällen können diese Ziele Zielkonflikte bedingen, da mehr Einfachheit nicht immer mit den Sicherheitsanforderungen vereinbar ist.
Mehrere etablierte Ressourcen unterstützen die formative Evaluierung während der frühen Entwicklungsphase der Geräte-Benutzeroberfläche. AAMI HE75, Human Factors Engineering – Design von MedizinproduktenAbschnitt 9 bietet praktische Hinweise zu Methoden des Usability-Testings wie Cognitive Walkthroughs, heuristischen Evaluationen und Walkthrough-Talkthrough-Studien. IEC 62366-1, Anhang D, beschreibt diese Techniken ebenfalls im Kontext des Usability Engineering.
Standards für Interface-Design und Best-Practice-Referenzen wie HE75 bieten hervorragende Leitlinien für die Gestaltung von Displays, Bedienelementen, grafischen Benutzeroberflächen, Alarmen, chirurgischen Instrumenten, Gebrauchsanweisungen und anderen UI-Elementen. HE75 betont jedoch die Bedeutung interaktiver Nutzertests während des UI-Designprozesses. Formative Techniken wie Cognitive Walkthroughs, heuristische Evaluationen und Walkthrough-Talkthrough-Usability-Tests sind unerlässlich, um dem Designteam umsetzbares Nutzerfeedback hinsichtlich Benutzerfreundlichkeit und effektiver Fehlerbehebung zu liefern.
Angemessene formative Evaluationen durchführen
Obwohl formative Evaluierungen in der Regel nur geringen Aufwand und Ressourcen erfordern, schränken viele Hersteller diese Aktivitäten während der Entwicklung von Benutzeroberflächen ein oder verzichten ganz darauf. Dies beruht häufig auf weit verbreiteten Missverständnissen, darunter den folgenden:
- Wahrgenommener Kosten- und Zeitaufwand: Formative Studien sind im Allgemeinen effizient und können mit einer kleinen Anzahl repräsentativer Nutzer durchgeführt werden. Werden sie früh im Designprozess eingesetzt, können Studien mit nur 8–10 Teilnehmern 90 % der bestehenden Designfehler aufdecken. Dies ermöglicht zeitnahe Designverbesserungen, die Fehler und nutzungsbedingte Probleme reduzieren. Mit zunehmender Reife des Designs können formative Evaluationen auch als Testläufe vor der abschließenden summativen Validierung dienen. Dadurch lassen sich kostspielige Überarbeitungen oder wiederholte Validierungen aufgrund ungelöster Usability-Probleme oder Protokollschwächen vermeiden.
- Verzögerungen bei der Einreichung von Unterlagen durch die Einholung der Genehmigung des Ethikkomitees: Viele Unternehmen glauben möglicherweise, dass formative Tests eine aufwendige Genehmigung durch eine Ethikkommission (Institutional Review Board, IRB) hinsichtlich der Teilnehmersicherheit erfordern. In den meisten Fällen bergen Usability-Tests jedoch kein Risiko für die Teilnehmer (d. h., es wird keine Therapie durchgeführt, da der Fokus auf der Interaktion der Nutzer mit der Benutzeroberfläche liegt). Hersteller können sich jedoch durch die Einholung einer IRB-Genehmigung vor Haftungsansprüchen schützen. Dies kann aufgrund der geringeren Komplexität der Tests im Vergleich zu klinischen Studien in Form von beschleunigten Prüfverfahren erfolgen.
- Annahmen zum Dokumentationsaufwand: Aus Sicht der FDA- und IEC 62366-1-Konformität erfordern formative Evaluierungen keine umfangreiche Dokumentation in den Zulassungsanträgen. Die FDA-Prüfer möchten lediglich wissen, ob der Hersteller iterative Tests am Produkt durchgeführt und alle Anstrengungen unternommen hat, sicherheitsrelevante Konstruktionsmängel zu beheben. In den letzten Jahren haben FDA-Prüfer jedoch zunehmend formative Evaluierungsberichte angefordert, um eine angemessene Vorbereitung vor der summativen Usability-Validierung nachzuweisen – eine Abkehr von früheren Anforderungen an die Zulassung.
Simulationstests (Validierung)
Summative Prüfungen, auch Validierungstests unter simulierten Anwendungsbedingungen genannt, sind die wichtigste Nachweisquelle für die Anwendungssicherheit gemäß IEC 62366-1 und den Richtlinien der FDA zu menschlichen Faktoren. Um offensichtliche Sicherheitsbedenken der Testteilnehmer zu vermeiden, können summative Prüfungen unter simulierten Anwendungsbedingungen durchgeführt werden, die realen Bedingungen entsprechen.
Summatives Testen unterscheidet sich von formativen Evaluationen hinsichtlich Zweck und Durchführung. Es ist kein Frontalunterricht. Diese Übung ist explorativ angelegt und zielt nicht auf die Einholung von Design-Input ab; vielmehr dient sie als abschließende Demonstration der sicheren und effektiven Anwendung des Geräts. Die Teilnehmer werden während der Aufgabenbearbeitung weder angeleitet, unterbrochen noch korrigiert.
Die Nutzungsszenarien sind so konzipiert, dass sie typische Interaktionsabläufe mit dem Gerät widerspiegeln und alle in der nutzungsbezogenen Risikoanalyse als risikoreich eingestuften Aufgaben umfassen. Schulung und Einarbeitung erfolgen unter Berücksichtigung der zu erwartenden realen Bedingungen, wobei Lernerfolg und mögliche Gedächtnislücken berücksichtigt werden. Die Gebrauchsanweisung wird während der Tests bereitgestellt, ist aber für die Teilnehmenden nicht verpflichtend.
Die Leistung der Nutzer wird beobachtet und als Erfolg, Misserfolg oder Erfolg mit Schwierigkeiten (z. B. Zögern, Selbstkorrektur oder Verwirrung) kategorisiert. Diese Beobachtungen sowie alle Misserfolge werden für die Nachanalyse dokumentiert. Im Anschluss an den Test werden Interviews durchgeführt, um die Ursachen der beobachteten Schwierigkeiten oder Fehler zu verstehen und Feedback der Teilnehmer zur Aufgabenkomplexität zu erhalten.
Die Studienergebnisse müssen eine Gesamtaussage zur Anwendungssicherheit ermöglichen. Diese Beurteilung basiert nicht auf vordefinierten quantitativen Erfolgsschwellenwerten, sondern darauf, ob verbleibende Muster anwendungsbezogener Probleme auf die Benutzeroberfläche, die Kennzeichnung oder die Gebrauchsanweisung zurückzuführen sind.
Gestaltung und Interpretation summativer Testergebnisse in Validierungsstudien mit simulierter Nutzung
Die Konzeption, Durchführung und Auswertung summativer Simulationstests gehören zu den anspruchsvollsten Aspekten der Mensch-Computer-Interaktion und Benutzerfreundlichkeitsforschung (Human Factors and Usability Engineering, HF/UE). Diese Studien liefern den wichtigsten Nachweis für die Benutzersicherheit und unterscheiden sich deutlich von herkömmlichen Usability-Tests. Ihr Wert liegt nicht nur in der beobachteten Aufgabenleistung, sondern auch in der anschließenden Analyse, die erforderlich ist, um die Ursachen von Anwendungsfehlern und -schwierigkeiten zu ermitteln.
Wie in den FDA-Richtlinien und IEC 62366-1 beschrieben, ermöglicht die summative Prüfung Herstellern eine vorausschauende Bewertung der tatsächlichen Geräteanwendung. Unerwartete oder unerwünschte Ergebnisse, die während der Prüfung beobachtet werden, sollten analog zur Analyse unerwünschter Ereignisse nach der Markteinführung untersucht werden. Daher werden unerwartete oder unerwünschte Beobachtungen in diesen Prüfungen genauso untersucht wie unerwünschte Ereignisse. Ziel ist es, die Ursachen von Fehlern bei kritischen Aufgaben zu identifizieren und festzustellen, ob diese Probleme vor der behördlichen Zulassung und Vermarktung behoben werden können.
Die Einreichung beim FDA-Zulassungsverfahren erforderte im Allgemeinen die Vorlage zusammenfassender Validierungstestergebnisse, insbesondere bei Medizinprodukten mit höherem Risiko. Obwohl sich die Studienqualität insgesamt im Laufe der Zeit verbessert hat, werden weiterhin mehrere wiederkehrende Mängel beobachtet:
- Unzureichende Verknüpfung mit der nutzerbezogenen Risikoanalyse (URRA): Testprotokolle sollten explizit mit Hilfe der URRA begründet werden (Figure 2Die Anwendungsszenarien werden aus Risikoperspektive kritischen Aufgaben zugeordnet. Kritische Aufgaben müssen klar dokumentiert und dürfen nicht implizit vom Prüfer abgeleitet werden. Eine übersichtliche Darstellung der Aufgabenanalyse, der Risikokontrollen und der Validierungsaktivitäten stärkt die regulatorische Prüfung.
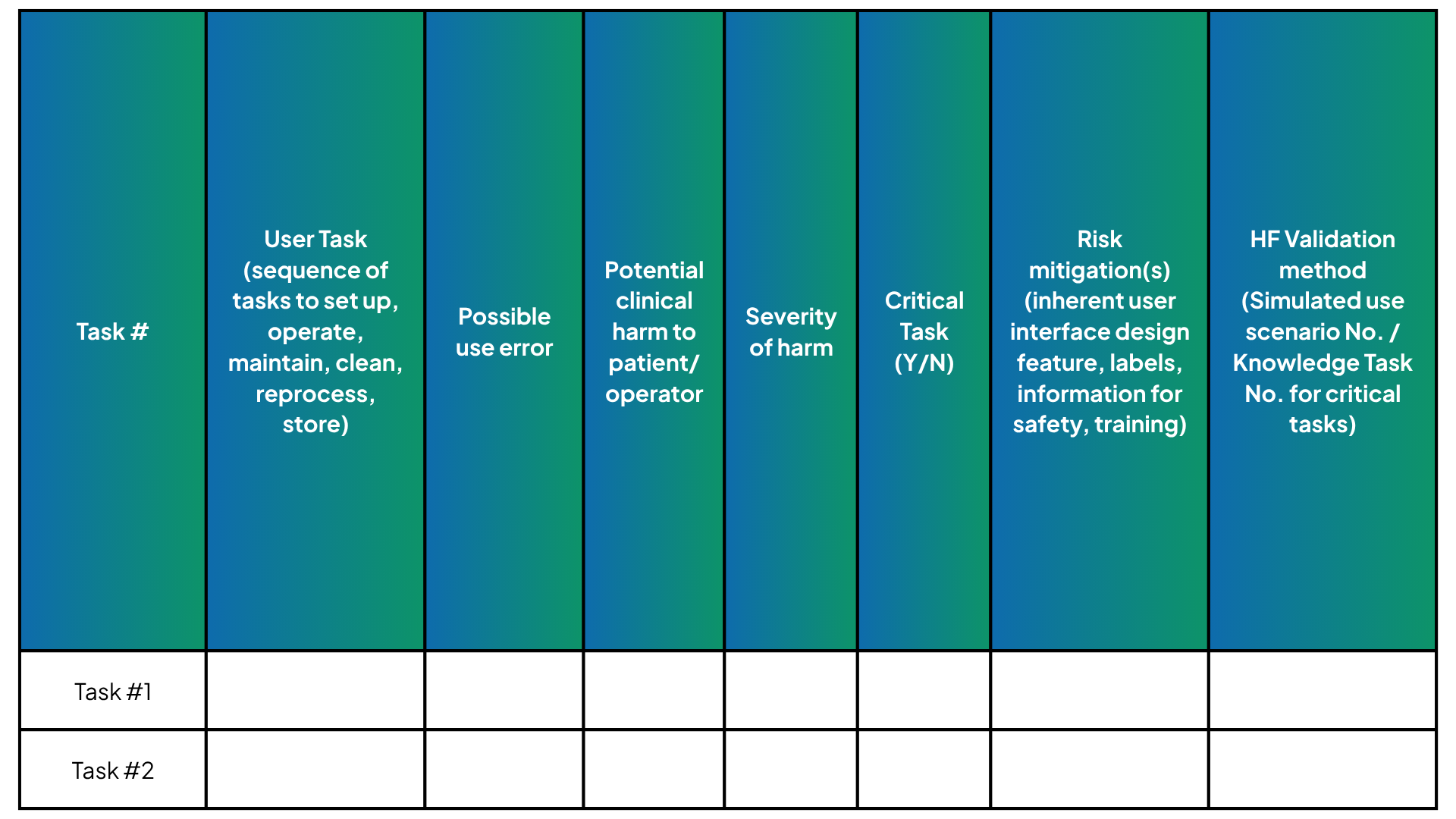
Abbildung 2: Beispiel für ein Aufgaben-, Risikokontroll- und Testschema
- Meldung von Erfolgsquoten ohne Gewährleistung der Anwendungssicherheit: Numerische Erfolgsquoten allein reichen nicht aus. Hersteller müssen Fehlerfälle auswerten und diskutieren, um festzustellen, ob selbst vereinzelte Anwendungsfehler plausiblerweise zu schwerwiegenden Patientenschäden führen könnten. Die Gesamtargumentation zur Anwendungssicherheit sollte auf Fehleranalysen und nicht auf Prozentzahlen basieren.
- Übermäßige Abhängigkeit von subjektiven Präferenzdaten: Bewertungen der Benutzerfreundlichkeit und Nutzerpräferenzen sind kein Beweis für eine sichere und korrekte Anwendung. Subjektive Kennzahlen können zwar Schlussfolgerungen stützen, ersetzen aber nicht die objektive Bewertung der Aufgabenleistung und der Fehlervermeidung. Die Annahme, das Gerät sei einfach zu bedienen, beweist weder die korrekte Anwendung noch liefert sie ein überzeugendes Argument gegenüber den Aufsichtsbehörden.
- Unzureichende oder nicht genügend Repräsentation von Nutzergruppen: Sowohl die FDA-Richtlinien als auch IEC 62366-1 empfehlen, mindestens 15 Teilnehmer pro identifizierter Nutzergruppe einzubeziehen. Bei der Bestimmung geeigneter Nutzergruppen sollten die ausführenden Personen, relevante Nutzerprofile sowie Unterschiede in Ausbildung, Erfahrung oder körperlichen bzw. kognitiven Fähigkeiten berücksichtigt werden. Geräte für den Heimgebrauch oder Kombinationsprodukte für die Anwendung durch Patienten können zusätzliche Nutzergruppen erfordern, beispielsweise Personen mit Seh- oder kognitiven Beeinträchtigungen. Es wird empfohlen, vor der Einreichung mit der FDA Rücksprache zu halten, um die Teststrategie abzustimmen.
- Keine Verzögerung zwischen Training und Test: Bei simulierten Anwendungstests sollte ein Zeitintervall zwischen Training und Aufgabenausführung eingehalten werden, um den Lernverlust zu berücksichtigen und reale Anwendungsbedingungen abzubilden. Bei einigen Zulassungsanträgen für Kombinationsprodukte kann die FDA eine geschulte und eine ungeschulte Gruppe verlangen.
- Fehler ohne Analyse auf „menschliches Versagen“ zurückführen: Die Ergebnisse der summativen Tests sollten fallweise analysiert werden, insbesondere im Hinblick auf beobachtete Fehler und Schwierigkeiten der Teilnehmenden. Die bloße Erklärung, ein Fehler sei auf ein „Vergessen“ während der Ausführung einer kritischen Aufgabe zurückzuführen, ist kein stichhaltiges Argument für die Sicherheit der Benutzeroberfläche. Diese sollte die Nutzenden während der Aufgabenbearbeitung bestmöglich unterstützen. Interviews nach dem Test sind unerlässlich, um die Denkweise der Nutzenden, mögliche Verwirrungen und deren Ursachen zu verstehen. Es ist absolut notwendig, die Teilnehmenden zu fragen, warum sie verwirrt waren, was sie zu der Annahme veranlasste, das Gerät funktioniere auf eine bestimmte Weise, oder warum sie einen Fehler gemacht haben.
- Mangelnde Überprüfung der Gebrauchsanweisung (IFU), wenn diese als primäre Risikokontrolle herangezogen wird: Wenn Hersteller die Gebrauchsanweisung als wichtige Risikominderungsmaßnahme einstufen, erwarten die Aufsichtsbehörden in der Regel gezielte Tests, um deren Wirksamkeit zu bestätigen. Die FDA-Leitlinien betonen, dass die alleinige Verwendung der Kennzeichnung im Allgemeinen nicht ausreicht und dass Sicherheitsmerkmale nach Möglichkeit in die Benutzeroberfläche integriert werden sollten.
Menschliche Faktoren und Usability-Tests als Wettbewerbsvorteil
Über die Einhaltung von Vorschriften hinaus bieten Human Factors und Usability-Tests einen strategischen Vorteil. Geräte, die intuitiv, effizient und auf reale Arbeitsabläufe abgestimmt sind, haben eine höhere Wahrscheinlichkeit:
- Von Klinikern übernommen werden
- Reduzierung des Schulungsaufwands
- Verbesserung der Nutzerzufriedenheit
- Probleme nach dem Börsengang minimieren
- Markenvertrauen stärken
In einem zunehmend wettbewerbsintensiven Medizintechnik-Umfeld ist Benutzerfreundlichkeit nicht nur eine regulatorische Anforderung, sondern ein Unterscheidungsmerkmal.
Fazit
Die Berücksichtigung menschlicher Faktoren und die Benutzerfreundlichkeit haben sich zu einem unverzichtbaren Bestandteil der Produktentwicklung entwickelt und gewährleisten die einfache und sichere Anwendung von Medizinprodukten. Weltweit fordern Aufsichtsbehörden ein systematisches Überwachungs- und Prüfverfahren hinsichtlich der Einhaltung der Norm für Benutzerfreundlichkeit (IEC 62366-1) durch die Hersteller. Das Zulassungsverfahren der FDA umfasst mittlerweile routinemäßig eine nutzerbezogene Risikoanalyse unter Berücksichtigung menschlicher Faktoren sowie Validierungstests der Benutzeroberfläche mit den vorgesehenen Anwendern.
HF/UE-Aktivitäten sollten in allen Phasen der Geräteentwicklung und -gestaltung durchgeführt werden, einschließlich der vorläufigen Aufgaben- und benutzerbezogenen Risikoanalyse, der UI-Gestaltung und -Bewertung sowie der abschließenden summativen Validierungstests in simulierter Nutzung.
Der HF/UE-Prozess sollte mit dem Gesamtrisikomanagementprozess für das Gerät abgestimmt sein. Endgültige Entscheidungen hinsichtlich der Akzeptanz des Nutzungsrisikos sollten auf den Ergebnissen von simulierten Nutzungstests mit repräsentativen Anwendern basieren, die das Vorhandensein verbleibender Muster sowie von Fehlern und Schwierigkeiten bei Aufgaben mit kritischen Risikoauswirkungen bewerten.
Formative und summative Human-Factors-Tests erfüllen unterschiedliche, aber sich ergänzende Funktionen in der Entwicklung von Medizinprodukten. Strategisch eingesetzt, ermöglichen sie Herstellern jeder Größe, sicherere Produkte zu entwickeln, regulatorische Prozesse zu vereinfachen und das Gesamtentwicklungsrisiko zu reduzieren.
- Formative Tests liefert die notwendigen Einblicke, um effektiv gestalten
- Summative Prüfung liefert die notwendigen Beweise Sicherheit demonstrieren
Zusammen bilden sie die Grundlage einer robusten, skalierbaren Strategie für Human Factors und Usability-Tests, die sowohl die Einhaltung von Vorschriften als auch den kommerziellen Erfolg unterstützt.
Häufig gestellte Fragen (FAQs)
Unterscheidet sich Benutzerfreundlichkeit von menschlichen Faktoren?
Der Begriff „Usability Engineering“, oft synonym mit „Human Factors“ verwendet, zielt darauf ab, Benutzeroberflächen so zu gestalten, dass sie schnelles Lernen, hohe Nutzerzufriedenheit und effiziente Interaktion ermöglichen. „Usability“ ist eine mehrdimensionale Eigenschaft, die die Fähigkeit eines Menschen beschreibt, einfach und möglichst fehlerfrei mit einem System oder Produkt zu interagieren.
Welche Normen oder regulatorischen Richtlinien sollten Sie im Hinblick auf die Gebrauchstauglichkeit von Medizinprodukten beachten?
Für die USA erkennt die FDA IEC 62366-1, Medizinprodukte – Teil 1: Anwendung der Usability-Technik auf Medizinprodukte, ANSI AAMI HE75, Human Factors Engineering – Design von Medizinprodukten und die FDA-Leitlinie „Anwendung von Human Factors und Usability Engineering auf Medizinprodukte“ vom Februar 2016 an.
Die internationale Regulierungsbehörde hat die Norm IEC 62366-1, Medizinprodukte – Teil 1: Anwendung der Gebrauchstauglichkeitstechnik auf Medizinprodukte, als Teil des Zulassungsverfahrens außerhalb der USA aufgenommen.
Sind Tests zur Ergonomie für alle Medizinprodukte erforderlich?
Das Zulassungsverfahren der FDA umfasst mittlerweile routinemäßig eine nutzerbezogene Risikoanalyse unter Berücksichtigung menschlicher Faktoren sowie Validierungstests der Benutzeroberfläche von Medizinprodukten mit den vorgesehenen Anwendern. Dies gilt insbesondere für Hochrisiko-Medizinprodukte und Software als Medizinprodukt (SaMD).
Wie viele Nutzer werden für formative Tests benötigt?
Für formative Evaluierungen ist keine feste oder Mindestanzahl an Nutzern erforderlich. Sowohl IEC 62366-1 als auch die FDA-Leitlinie „Applying Human Factors and Usability Engineering to Medical Devices“ betonen, dass formative Tests iterativ und explorativ sind und darauf abzielen, Usability-Probleme und nutzungsbezogene Risiken zu identifizieren – nicht die Leistung statistisch zu validieren.
Obwohl die Regulierungsbehörden keine bestimmte Stichprobengröße vorschreiben, haben viele Hersteller festgestellt, dass 8-10 repräsentative Benutzer 90 % der vorhandenen Konstruktionsfehler aufdecken können.
Worin unterscheidet sich summatives Testen von anderen Usability-Tests?
Es bestehen einige wichtige Unterschiede in der Testmethodik zwischen summativen simulierten Nutzungstests und anderen Usability-Bewertungen wie formativen Tests. Der summative Test dient nicht der explorativen Suche nach Feedback zu Designmerkmalen, sondern wird mit dem finalen Gerätedesign durchgeführt. Er umfasst repräsentative Nutzer, Umgebungen und Nutzungsszenarien und folgt einem strukturierten, vordefinierten Protokoll, das sich auf kritische Aufgaben und Worst-Case-Szenarien konzentriert. Die Ergebnisse des summativen Tests werden in einem formalen Bericht zur Einreichung bei den Aufsichtsbehörden dokumentiert.
Kann ein intensives formatives Testen das Risiko eines Scheiterns bei summativen Tests verringern?
Ja. Iteratives formatives Testen erhöht die Wahrscheinlichkeit einer erfolgreichen summativen Validierung und einer reibungsloseren behördlichen Prüfung deutlich.
Ressourcen und Referenzen
FDA-Leitfaden für die Industrie und Mitarbeiter der Lebensmittel- und Arzneimittelbehörde – Anwendung von Human Factors und Usability Engineering auf Medizinprodukte, veröffentlicht am 3. Februar 2016
ANSI AAMI HE75:2009/(R)2018, Human Factors Engineering – Design of Medical Devices (FDA-anerkannt Nr. 5-57)
IEC 62366-1 Ausgabe 1.1 2020-06 Konsolidierte Fassung, Medizinprodukte – Teil 1: Anwendung der Usability-Engineering-Methode auf Medizinprodukte (FDA-anerkannt Nr. 5-129)
ISO 14971 Dritte Ausgabe 2019-12, Medizinprodukte – Anwendung des Risikomanagements auf Medizinprodukte (FDA-anerkannt Nr. 5-125)
Wiklund, J. Kendler und A. Strochlic, Usability Testing of Medical Devices. CRC Press-Taylor & Francis Group (2011)
