PMCF-Pläne, Umfragen und Berichte für Medizinprodukte
Wenn Sie Geräte in Europa vermarkten, wissen Sie bereits, dass Ihre Pflichten zur klinischen Bewertung gemäß der EU-Medizinprodukteverordnung nicht mit der CE-Kennzeichnung enden. Die Medizinprodukteverordnung (MDR) verlangt von Ihnen, proaktiv Daten zu sammeln, um die fortlaufende Sicherheit und Leistung der Geräte zu bewerten. PMCF, auch bekannt als Post Market Clinical Follow-up, ist ein wichtiger Teil Ihrer gesamten Post Market Surveillance (PMS)-Aktivitäten. NAMSA kann Ihnen dabei helfen, eine Strategie zu entwickeln, welche Daten wie erfasst werden sollen, und einen aussagekräftigen PMCF-Bericht vorzubereiten, der die Erwartungen Ihrer Benannten Stelle erfüllt.
Kurzinfos zur klinischen Nachbeobachtung nach der Markteinführung:
- Erforderlich in Artikel 61 und Anhang XIV (Teil B) der europäischen MDR (2017/745)
- Kritische Komponente der Aktivitäten zur Überwachung nach dem Inverkehrbringen
- Gilt für alle Klassen von Medizinprodukten, auch für solche mit einer langen und sicheren Leistungsgeschichte
- Der PMCF-Bericht wird Teil Ihres Clinical Evaluation Report (CER)
- Gilt für den gesamten Lebenszyklus des Medizinprodukts
- Intensität des PMS proportional zur Risikoklasse und Art des Geräts
Wie NAMSA Kann Ihnen helfen, die PMCF-Anforderungen zu erfüllen
Die klinische Nachbeobachtung nach der Markteinführung ist ein systematischer Prozess und besteht aus mehreren Phasen, wie unten dargestellt.
Schritt 1: Wir helfen Ihnen bei der Definition Ihrer Strategie
Wir arbeiten mit unseren Kunden zusammen, um eine solide PMCF-Strategie zu entwickeln, bevor wir direkt in den Planungsmodus einsteigen. Warum? Um die MDR-Anforderungen (und Ihre Benannte Stelle) zu erfüllen, ist es wichtig, dass Sie die richtigen Daten auf die richtige Weise erfassen. Um eine Strategie zu entwickeln, NAMSA wird mehrere Faktoren bewerten, darunter Kosten, Praktikabilität, Zeitpläne, Anzahl der vermarkteten Geräte, wissenschaftliche Gründe und Geschäftsentscheidungen. Wir werden eine Strategie mit Input von Ihnen und unseren klinischen und regulatorischen Teams entwerfen. Sobald die Strategie vereinbart ist, verwenden wir sie, um Entscheidungen über Ihren PMCF-Plan zu treffen.
Schritt 2: Erstellen Sie anhand Ihrer Strategie einen PMCF-Plan für Medizinprodukte
Der PMCF-Plan ist ein Dokument, in dem beschrieben wird, welche PMCF-Aktivitäten Sie für ein bestimmtes Gerät durchführen möchten. MDCG 2020-7 bietet eine täuschend einfache Vorlage zum Erstellen eines PMCF-Plans. Diese PMCF-Vorlage sagt Ihnen, was erforderlich ist, aber nicht, wie Sie es tun oder was Ihre benannte Stelle von Ihnen erwartet. Unsere internen klinischen und regulatorischen Teams haben Hersteller medizinischer Geräte weltweit bei der klinischen Nachverfolgung nach der Markteinführung unterstützt und entwickeln einen umfassenden PMCF-Plan, der die Methoden und Verfahren angibt, die Sie zum Sammeln von Daten verwenden werden, und wie Sie:
- Bestätigen Sie die fortlaufende Sicherheit und Leistung Ihres Medizinprodukts
- Identifizieren Sie unbekannte Nebenwirkungen, neu auftretende Risiken, Off-Label-Anwendung oder systematischen Missbrauch
- Stellen Sie sicher, dass das in Ihrem Risikomanagementplan festgelegte Nutzen-Risiko-Verhältnis noch akzeptabel ist
In den meisten Fällen sind klinische Studien nach der Markteinführung nicht erforderlich, wenn ausreichend Daten verfügbar sind und die von Ihrem Produkt ausgehenden Risiken gering sind. Normalerweise können andere Aktivitäten wie Literaturrecherchen und Umfragen ausreichend sein. Wenn dies der Fall ist, begründen wir dies in Ihrem PMCF-Plan.
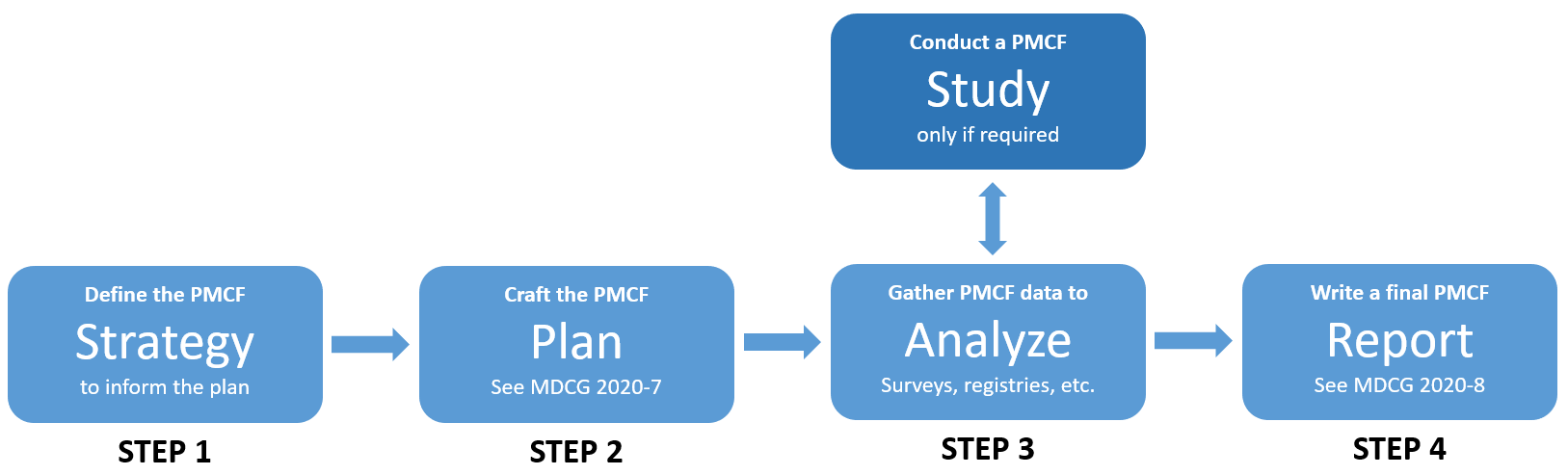
Schritt 3: Sammeln Sie Daten über PMCF-Umfragen, Register und mehr, wie in Ihrem Plan angegeben
Obwohl die EU-MDR einige Ausnahmen zulässt, müssen für die meisten Geräte kontinuierlich PMCF-Daten erhoben werden. Post-Market-Umfragen sind eine der am häufigsten verwendeten Methoden zur Erhebung von Daten von Benutzern und Patienten. NAMSA verfügt über umfassende Erfahrung in der Entwicklung, Durchführung und Analyse von PMCF-Umfragen sowie in vielen anderen Quellen für Post-Market-Daten, wie unten aufgeführt. Unser Team hilft Ihnen beim Sammeln neuer PMCF-Daten und überprüft vorhandene Daten, die Ihnen aus folgenden Quellen zur Verfügung stehen:
- Hochwertige klinische Studien
- Fokusgruppen
- Einzelne Fallberichte
- Usability-Befragungen inklusive Arzt- oder Patientenfeedback
- Feedback von Vertriebs- oder Account-Managern
- Veröffentlicht Kataloge auf dem aktuellen Stand der Technik oder gleichwertigen Geräten
- Klinische Registerstudien
- Vollständige oder teilweise klinische Studien nach der Markteinführung
Durchführen einer PMCF-Studie für Medizinprodukte
Die Tiefe und Breite der erhobenen Daten hängt weitgehend von der Risikoklasse Ihres Produkts ab. Wenn die mittel- oder langfristige Sicherheit und klinische Leistung Ihres Produkts durch praktische Erfahrungen gut dokumentiert ist, ist eine klinische Studie nach der Markteinführung möglicherweise nicht erforderlich. Wenn jedoch ein PMCF-Studie Wird benötigt, NAMSA verfügt über in den USA und Europa ansässige Teams, die Ihre Studie planen und verwalten können, einschließlich Protokollentwicklung, Studienmanagement, Patienten-/Benutzerrekrutierung, Biostatistik und mehr.
Schritt 4: Schreiben Sie den PMCF-Evaluierungsbericht
Nachdem alle Daten erhoben und ausgewertet wurden, ist es an der Zeit, den Bericht zu schreiben. Auch dafür gibt es eine Vorlage! MDCG 2020-8 bietet einen grundlegenden Rahmen dafür, was enthalten sein sollte. Denn NAMSA hat an so vielen PMCF-Projekten mit einer großen Bandbreite an Geräten gearbeitet, dass wir ein sehr gutes Gespür dafür haben, wonach benannte Stellen suchen. Daten aus dem Bericht werden verwendet, um Ihren klinischen Bewertungsbericht, Ihre Risikomanagementdatei, Ihre Zusammenfassung der Sicherheit und klinischen Leistung usw. zu aktualisieren. Da wir wiederum eines der größten CROs für Medizinprodukte sind, verfügen wir über die interne Expertise, um alle Aspekte Ihres PMCF-Projekts abzuwickeln, von der Strategie über die Studie bis hin zur zusammenfassenden Berichterstattung.
Vereinbaren Sie ein Treffen mit einem NAMSA PMCF-Experte
Häufig gestellte Fragen zu PMCF
PMCF gilt für alle in Europa verkauften Medizinprodukte. Kurz gesagt handelt es sich bei PMCF um den fortlaufenden Prozess der Aktualisierung des Berichts zur klinischen Bewertung (CER), der ursprünglich erstellt und überprüft wurde, bevor das Produkt auf den EU-Markt gebracht wurde. Gemäß der EU-Medizinprodukteverordnung (2017/745) endet die klinische Bewertung nicht, sobald das Produkt die CE-Kennzeichnung erhält – PMCF muss während des gesamten Lebenszyklus des Produkts durchgeführt werden. Die Anforderung zur Durchführung von PMCF ist in Artikel 61 und Teil A des Anhangs XIV der MDR vorgeschrieben.
PMCF ist immer für alle Klassen von Medizinprodukten erforderlich, auch für selbstzertifizierte Geräte der Klasse 1. Es spielt keine Rolle, ob Ihr Gerät in Europa seit Jahrzehnten sicher verwendet wird – Sie müssen proaktiv und regelmäßig Beweise dafür sammeln, dass das Gerät weiterhin sicher verwendet wird und wirksam ist. Die Intensität, mit der Sie PMCF angehen, variiert je nach Geräteklassifizierung erheblich, und Ihr Plan zum Sammeln und Analysieren von Daten ist in Ihrem PMCF-Plan dokumentiert.
Zu den Umständen, die PMCF-Studien rechtfertigen können, zählen unter anderem: Innovationen (neues Material, neue Technologie, neue Indikation usw.), wesentliche Änderungen am Gerät oder seinem Verwendungszweck, Invasivität des Verfahrens, anatomische Hochrisikostellen, Hochrisikopopulationen, die Schwere der behandelten Erkrankung, hohe erforderliche Fähigkeiten zur Verwendung des Geräts und Reproduktion der klinischen Ergebnisse, unbeantwortete Fragen zur langfristigen Sicherheit und Leistung oder die Identifizierung einer Untergruppe mit möglicherweise anderem Risiko-Nutzen-Verhältnis.
Auch für Altgeräte (Geräte, die sich bereits unter der MDD auf dem Markt befinden) können PMCF-Studien erforderlich sein, um die Anforderungen der MDR zu erfüllen.
Lernen Sie unser Team aus MDR-Regulierungsexperten und medizinischen Autoren kennen
-
Kevin Butcher
 Leitender RegulierungsberaterBiografie anzeigen
Leitender RegulierungsberaterBiografie anzeigen -
Jane Arnold-Round, MSc
 Leitender Hauptberater, RegulierungBiografie anzeigen
Leitender Hauptberater, RegulierungBiografie anzeigen -
Matt Royle, PhD
 Leitender RegulierungsberaterBiografie anzeigen
Leitender RegulierungsberaterBiografie anzeigen -
Rachel Gibbs, BSc, PhD
 Leitender RegulierungsberaterBiografie anzeigen
Leitender RegulierungsberaterBiografie anzeigen -
Thomas Miramond, PhD, MSc, M-Eng.
 Leitender RegulierungsberaterBiografie anzeigen
Leitender RegulierungsberaterBiografie anzeigen
Ähnliche Dienstleistungen, die Sie interessieren könnten
Globale RA/QA-Beratung

FSP-Outsourcing
